MEDUSA input and output
MEDUSA takes as input an amino acid sequence in .fasta format. Using HHblits tool, it extracts evolutionary information and combines it with physico-chemical properties of each residue to predict a flexibility class of each position using a convolutional neural network.
MEDUSA performs four different predictions in terms of the normalized B-factor value (Bnorm). It creates two 2-class predictions following the nomenclature introduced by Schlessinger & Rost in 2006 [1]:
- S (strict) two-class prediction with threshold Bnorm = 0.03
- NS (non-strict) two-class prediction with threshold Bnorm = -0.3
and multi-class predictions:
- Three-class prediction with following class limits: [-4, -0.5), [-0.5, 1), [1, ∞)
- Five-class prediction with following class limits: [-4, -1), [-1, 0), [0, 1), [1, 2), [2, ∞)
MEDUSA main algorithm
The general MEDUSA workflow is given in the figure below:
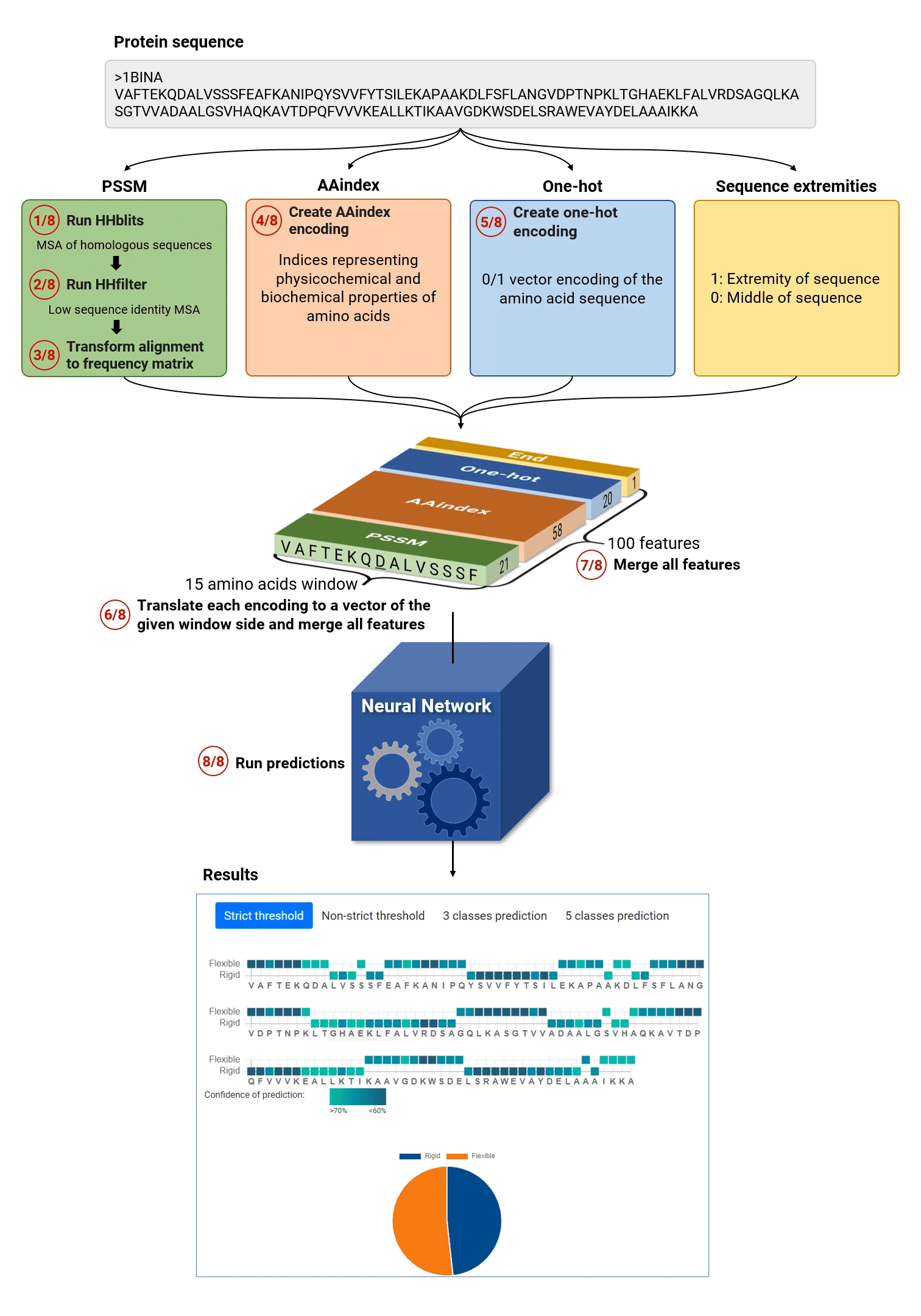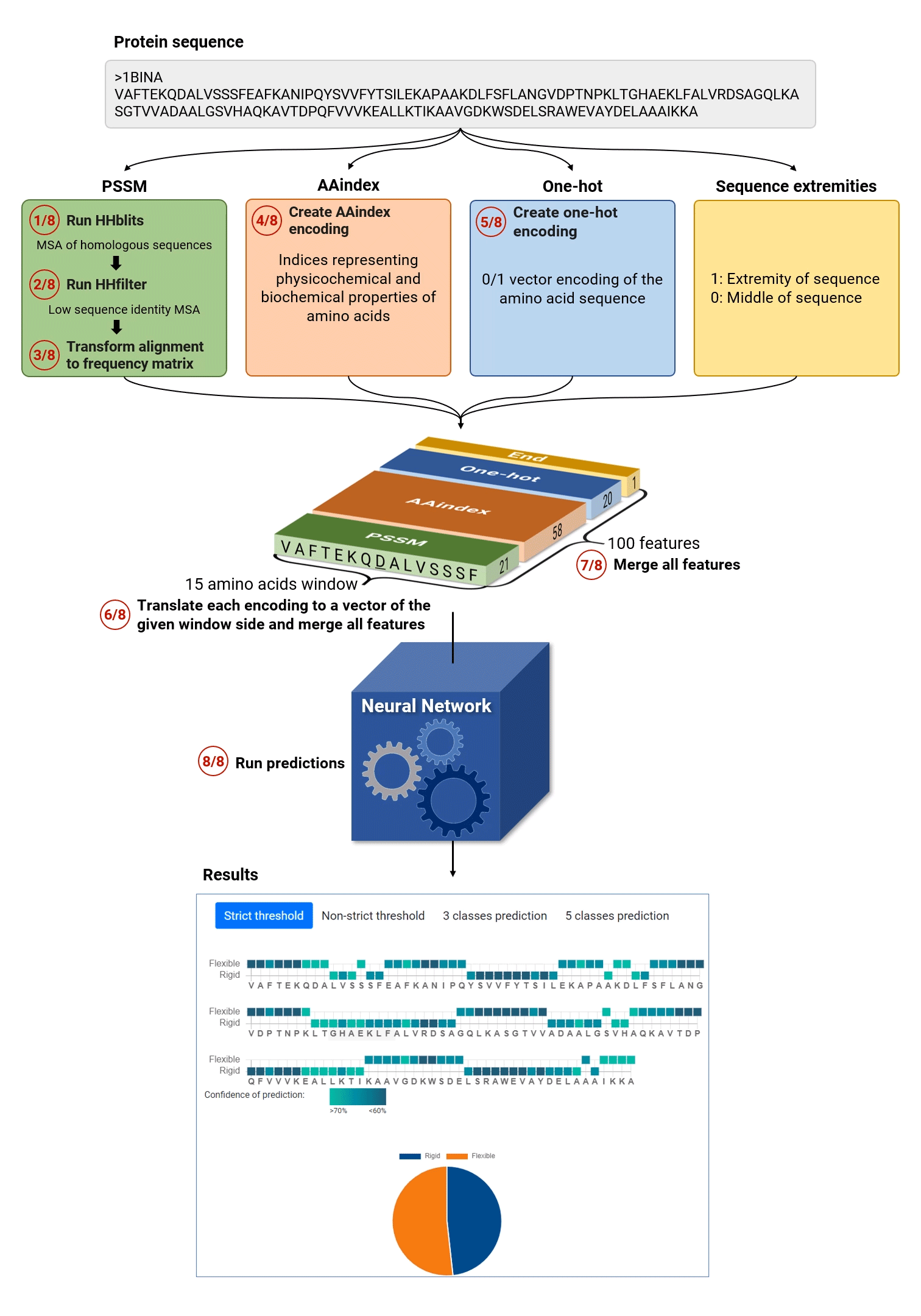
MEDUSA algorithm proceeds in eight main steps:
- Extract evolutionary information: MEDUSA finds homologs of the query sequence by HHblits search.
- MEDUSA filters the resulting Multiple sequence alignment (MSA) file using hhfilter
- The final MSA is translated into a probability profile using position specific score matrix: each position of the sequence is thus encoded by 21 numerical values corresponding to 20 amino acid types and gap.
- MEDUSA translates each amino acid to 58 numerical values, which encode its physico-chemical properties (using AA INDEX scheme).
- MEDUSA creates one hot encoding of each amino acid and adds a flag for the sequence terminus.
- Using a sliding window of 15 amino acids, MEDUSA creates input vectors for each sequence position for all the considered features.
- Different features are merged to create an input vector for the prediction of dimensions 15x100.
- Finally, the neural network performs binary and multi-class predictions and provides the general summary as well as flexibility prediction and confidence value for each amino acid.