SWORD (SWift and Optimized Recognition of structural Domains) is an automated
method that identifies protein domains using information on protein internal contacts between the residues.
For a given protein structure, SWORD can provide multiple alternative decompositions into domains.
To have more informations on methods implemented in SWORD2 please look at:
Postic G, Ghouzam Y, Chebrek R, Gelly J-C. Science Advances (2017) 3:e1600552
Gelly J-C, Lin H-Y, de Brevern AG, Chuang T-J, Chen F-C. Genome Biol Evol (2012) 4:966-975
Gelly J-C, de Brevern AG. Bioinformatics (2011) 27(1):132-3
Gelly J-C, Etchebest C, Hazout S, de Brevern AG. Nucleic Acids Res (2006)
Gelly J-C, de Brevern AG, Hazout S. Bioinformatics (2006) 22:129-133
SWORD2 main algorithm
SWORD takes as input a protein structure in .pdb format (can be either a PDB id from the PDB database, or proprietary structure or ID from the AlphaFold Protein Structure Database) and performs protein domain assignment using a partitioning algorithm based on hierarchical clustering of Protein Units. The web-server implements a method published in 1,2,3,4.
Protein Units (PUs) are evolutionarily preserved protein substructures describing the protein architecture at an intermediate level between secondary structures and domains (1,2,3,4). SWORD identifies PUs using Protein Peeling algorithm (4), which translates protein Cα-Cα distances into a distance probability matrix and performs its dissection by optimising 'partition index' reflecting structural independence of the subunits. The details and evolution of the method are provided in (2,3,4) and in the corresponding web service: Peeling 3.
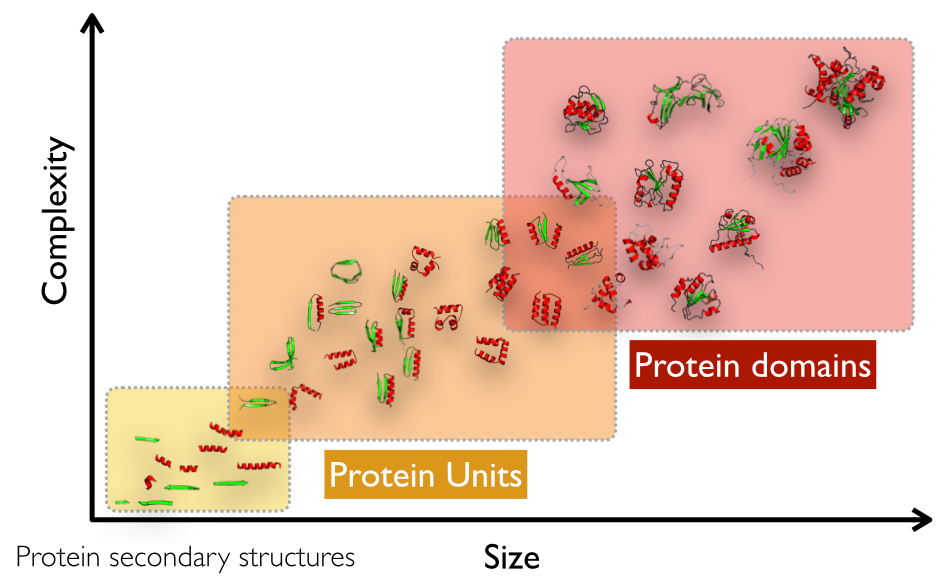
After PU identification SWORD gradually merges PUs and finds optimal domain delineation using two criteria: the separation (σ) and the compactness (κ). High value of σi,j indicates a high number of contacts between PUs i and j, meaning that these PUs are good candidates to be merged. Compactness criterion κi,j measures the contact density of the protein domain resulting from merging PUs i and j, and thus its high value that merging of PUs is favourable (1).
The optimal domain assignment is chosen according to the highest domain compactness. We also assess the quality of assignment using an estimation as a function of the Euclidean distance between the decomposition i and the threshold of the acceptance region. Then, for each structure we also report a measure of structural ambiguity, called the A-index which is introduced in (1). This measure is similar to the Hirsch index (h-index) used in scientometrics, except that it is based on the decomposition quality. Thus, a protein structure with an A-index of 3 has at least three different decompositions, each with a quality of 3 (The full description of the method is provided in (1).
Finally, the structural units, i.e. structural domains and Protein Units are assessed using the scoring function TIG-score Total Information Gain score (5) which allows to predict the native-like character of a 3D structure. This score is designed to behave like the Gibbs free energy and is often referred to as "pseudo-energy".
Based on pairwise distances, the value of the TIG tends to increase with increasing macromolecule size. To overcome this bias, a Z-score of the TIG is computed, following the atom shuffling method (7): for each domain, 2000 random sequence decoys are generated, the pseudo-energies of which follow a non-normal distribution of parameters μ and σ. The Z-score is calculated as (E − μ)/σ and, therefore, expresses the distance (in standard deviations σ) between the pseudo-energy of the domain E and those of the random decoys. To provide users a direct interpretation of the Z-score value, SWORD2 outputs the probability estimated by Chebyshev's inequality that we call Autonomous Unit Likelihood (AUL). Thus, for a substructure with a Z-score of −2.0, the probability of not observing the same pseudo-energy for a randomly delineated domain reaches 75%. This probability of being native reaches 94% for a domain with a Z-score of −4.0. The higher the probability, the more likely it is that the region under consideration is capable of autonomous folding.
SWORD2 Output
SWORD provides both a user-friendly interface for visualisation of the dissection results and raw output files available for download in the form of .tar.gz archive.
The output folder contains following files:
results/
SWORD2_summary.txt: summary of SWORD2 domain optimal and alternative assignments, their corresponding Protein Units as well as the AUL and Z-score for each element.SWORD2_summary.json: same informations as inSWORD2_summary.txtbut in easily parseable JSON format.mapping_auth_resnums.txt: file mapping the author residue numbers with the new numbering used on the results page.PDB_Name.pdb: initial structure submitted to the server if uploaded by the user, or the original PDB file corresponding to the 4 letters code submitted by the user.PDB_Name_ChainID.pdb: structure after cleaning by intermediate scripts (only the specified chain is written while keeping only the 20 standard amino acids).PDB_Name_ChainID.dssp: secondary structure assignment by DSSP for the considered protein.PDB_Name_ChainID_dssp.pdb: the cleaned structure to which was added as a header the DSSP secondary structure assignments.contact_probability_maps/: heatmaps of distance probability matrices for each domain partitioning as well as for each domain individually.SWORD/: PUs identified by Protein Peeling algorithm (Peeling subdirectory) and raw SWORD output for the optimal solution.SWORD/sword.txt: summary file of the SWORD output containing protein structure ambiguity (A-index, as described above) and protein partition in domains for optimal delineation (ASSIGNMENT) and all the alternatives (ALTERNATIVES). For each partition we report a number of domains (#D), minimal domain size (Min), limits of each domain (separated by a space, if a domain contains two PUs, they are separated by semicolons), the average compactness of the resulting partition (κ) and its quality.Protein_Units/: result files of the Peeling algorithm. For each Protein Unit determined, the following files are given: corresponding fasta (.fasta), secondary structures (.s2d), full PDB file (.pdb) and coordinates of Calpha residues only (.ca).Junctions/: TSV formated file summarizing the consistency of the different hinges/junctions. "Jnct" is the residue number corresponding to the extremity of a Protein Units/Domain determined by SWORD. "Cnt" is the number of times it found among all SWORD alternative partitionings. "Raw" is the raw frequency, standardized by the total number of domain delineations produced. "Wei" represents the frequency weighted by delineation quality and standardised by the total number of domain delineations produced.
logs/
JOB_ID_parameters.txt: parameters submitted by the user to the webserver.JOB_ID.log: informations about intermediate processing.
References
1. Postic G, Ghouzam Y, Chebrek R, Gelly J-C. An ambiguity principle for assigning protein structural domains. Science Advances (2017)3:e1600552
2. Gelly JC, de Brevern AG. Protein Peeling 3D: new tools for analyzing protein structures. Bioinformatics. 2011 Jan 1;27(1):132-3.
3. Gelly JC, Etchebest C, Hazout S, de Brevern AG. (2006) Protein Peeling 2: a web server to convert protein structures into series of protein units. Nucleic Acids Res. Jul 1;34 (Web Server issue):W75-8
4. Gelly JC, de Brevern AG, Hazout S. (2006) 'Protein Peeling': an approach for splitting a 3D protein structure into compact fragments. Bioinformatics. Jan 15;22(2):129-33. Epub 2005 Nov 14.
5. Postic G, Janel N, Tufféry P, Moroy G. An information gain-based approach for evaluating protein structure models. Comput Struct Biotechnol J. 2020;18:2228-2236.
6. Gelly J-C, Lin H-Y, de Brevern AG, Chuang T-J, Chen F-C. Selective Constraint on Human Pre-mRNA Splicing by Protein Structural Properties. Genome Biol Evol (2012)4:966-975
7. Bowie J.U., Lüthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991; 253:164–170.
Browser compatibilities
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari | Opera |
|---|---|---|---|---|---|---|
| Linux | Ubuntu 20.04 | 96.0.4664.110 | 95.0 | 96.0.1054.62 | n/a | 82.0.4227.33 |
| macOS | Monterey | 102 | 102 | n/a | 15.4 | 82.0.4227.43 |
| Windows | 10, 11 | 96.0.4664.110 | 95.0.1 | 96.0.1054.62 | n/a | 82.0.4227.33 |