Tools
The tools presented below redirect to their web-server versions hosted
either on our server (at Necker Hospital) or a third-party server (University or partner lab).
STRUCTURE ANALYSIS
SWORD2
SWift and Optimized Recognition of protein Domains
An automated method that identifies protein domains using information on protein internal contacts between the residues.
MEDUSA
Multiclass flexibility prediction from sequences of amino acids
MEDUSA is a Deep Learning approach for prediction of protein flexibility from sequence.
Protein Peeling 3
Peel a protein
An approach for splitting a 3D protein structure into compact fragments.
SWORD
SWift and Optimized Recognition
of structural Domains
A partitioning algorithm capable of producing multiple alternative domain assignments for a given protein structure.

OREMPRO
ORientation and Evaluation
of Membrane PROteins
A tool for the structural assessment of protein transmembrane domains.
Protein Block Expert
Web-based protein structure analysis server using a structural alphabet
PBE server 2.0 aims to provide a platform for protein structure analysis and comparison using well defined library of short structural motifs (SSMs) known as structural alphabets (SA).
VLDP
Determine protein contacts, accessibility and residue volume using Laguerre diagram
VLDPws is a tool for analysing protein structures based on Laguerre diagram, a powerful mathematical-geometric method.
STRUCTURE PREDICTION

ORION
Optimized protein fold RecognitION
ORION is a new profile-profile fold recognition approach that relies on a better description of the local protein structure to boost distantly protein detection.
PredyFlexy
Flexibility and Local Structure prediction from sequence
PredyFlexy predicts from the sequence: (i) the series of Long Structural Prototypes (LSPs), (ii) a sophisticated structural alphabet and (iii) the protein flexibility, this last being defined from data coming from X-ray structures and molecular dynamics simulation.
PB-kPRED
Knowledge-based prediction of
local structures using protein blocks
Knowledge-based algorithm to predict most probable backbone conformations for the protein local structures in terms of Protein Blocks.
COUDES
A tool for predicting beta-turns and their type in a protein or peptide sequence.
The basic principle is to predict first secondary structures with an accurate method such as PSIPRED (Jones, 1999), and to predict beta-turns within (almost) coil regions.

LocPred
A tool for local structure prediction from protein sequence using a structural alphabet approach
LocPred is a software which allows the users to submit a protein sequence and performs a prediction in terms of PBs.
STRUCTURE COMPARISON
iPBA
Protein Block Alignment
A tool for protein structures mining and superimposition based on similarity in the local backbone conformation.

mulPBA
Multiple Protein Block Alignment
mulPBA is an efficient tool for comparison of protein structures based on similarity in the local backbone conformation.
STRUCTURE ANALYSIS
SWORD2
SWift and Optimized Recognition of protein Domains
An automated method that identifies protein domains using information on protein internal contacts between the residues.
PYTHIA
Deep Learning Approach For Local Protein Conformation Prediction
Prediction of the protein local conformations in terms of PBs directly from the amino acid sequence.
MAIDEN
Model quality Assessment for Intramembrane Domains using an ENergy criterion
A statistical potential optimized on native alpha-helica and beta-sheet membrane protein structures.
ORION
Optimized protein fold RecognitION
ORION is a new profile-profile fold recognition approach that relies on a better description of the local protein structure to boost distantly protein detection.
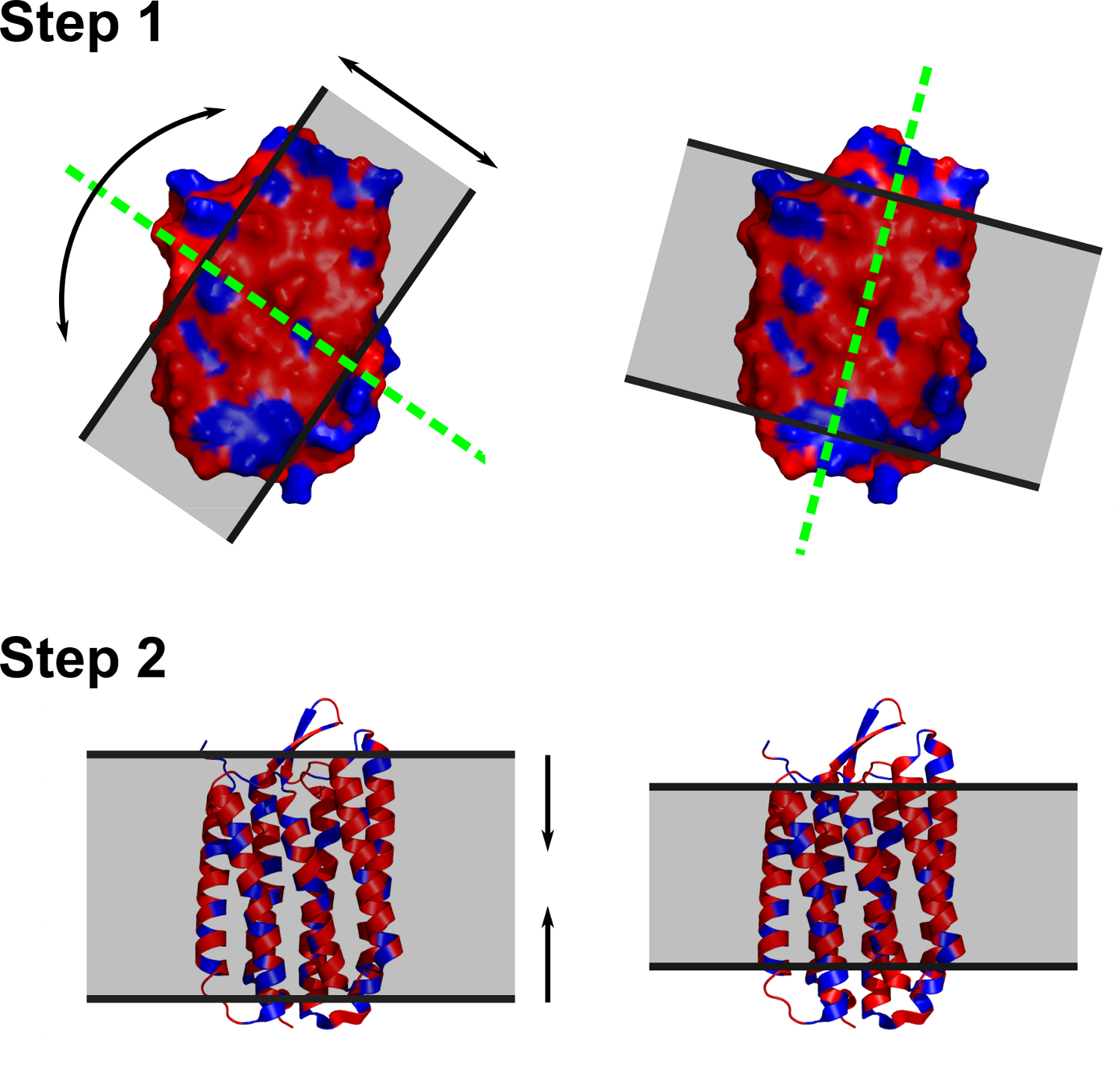
ANVIL
Assignment aNd VIsualization of the Lipid bilayer
ANVIL is aimed at assigning membrane boundaries to a protein 3D structure, by using the spatial coordinates of the alpha carbons.
KAKSI
Secondary Structure Assignment
KAKSI is a software dedicated to the assignment of classical repetitive secondary structure.
STRUCTURE PREDICTION
PredyFlexy
Flexibility and Local Structure prediction from sequence
PredyFlexy predicts from the sequence: (i) the series of Long Structural Prototypes (LSPs), (ii) a sophisticated structural alphabet and (iii) the protein flexibility, this last being defined from data coming from X-ray structures and molecular dynamics simulation.
SEQUENCE ANALYSIS
PoincaréMSA
Poincaré maps for visualization of large protein famillies
PoincaréMSA builds an interactive projection of an input protein multiple sequence alignemnt (MSA) using a method based on Poincaré maps.
SPECIALIZED DATABASES
ATLAS
Atlas of protein molecular dynamics
ATLAS gathers standardized molecular dynamics simulations of protein structures accompanied by their analysis in the form of interactive diagrams and trajectory visualisation. All the raw trajectories as well as the results of analysis are available for download.
RHeference
Database dedicated to RH alleles, focusing on RHD alleles and RHCE alleles expressing D epitopes
RHeference provides an overview of current knowledge regarding these alleles and an easy access to all relevant information for each allele.

CALR-ETdb
Calreticulin variant database involved in essential thrombocythemia
This project aims to compile all the variants of Calreticulin involved in essential thrombocythemia within the same database, namely CALR-ETdb.

RESPIRE
Repository of Enhanced Structures of Proteins Involved in the Red blood cell Environment
This database aims at providing a reference, ordered and protein-driven information on proteins available in the erythrocyte.
PolyprOnline
A database dedicated to Polyproline II Helix conformations in proteins
PolyprOnline is a requestable database dedicated to the assignment of PolyProline II helices.
PTM-SD
Post Translational Modification Structural Database
PTM-SD provides an access to proteins for which Post Translational Modifications are both experimentally annotated and structurally resolved.
MitoGenesisDB
Mitochondrial Spatio-Temporal Expression Database
The database MitoGenesisDB focuses on the dynamic of mitochondrial protein formation through global mRNA analyses.
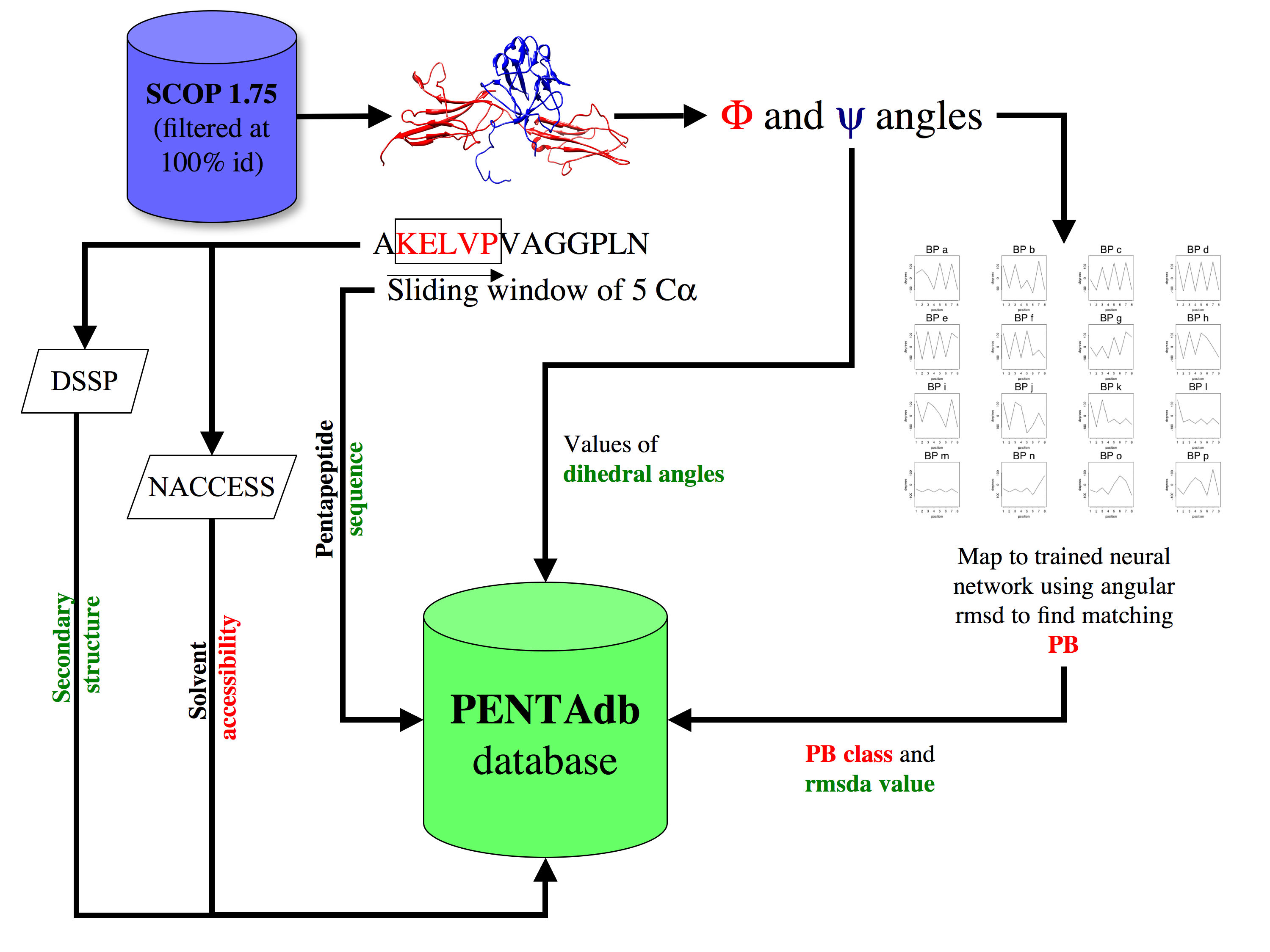
PB-PENTAdb
Database of pentapeptides from protein structures
A platform to investigate the structural features of pentapeptides in protein structures.
KNOTTIN
KNOTTIN database
Knottins are small disulfide-rich proteins characterized by a very special "disulfide through disulfide knot"
