The Red Blood Cell
The erythrocyte, also called Red Blood Cell (RBC) differenciates into the simplest human cell during the process of erythropoiesis. The lost of nuclei and internal organelles transform the multipotent haematopoietic cell into a simplified but flexible lipid compartment made of and containing a lot of proteins. With a production by the organism of up to 2.5 millions erythrocytes per second, this cell is also one of the easiest to extract and study.
Information
Our database aims at providing a comprehensive structural view of the Red Blood Cell protein content. The protein list was initially assembled from published articles, mostly coming from massive proteomics analyses of the RBC content by experts. These proteomics studies have provided important insights into the protein composition and organisation inside the RBC, but an integrated database presenting these data was not available. Our database aims at integrating more large-scale studies as new massive data become availabe, to define the hallmark for data integration for blood-derived cells. This unique combination allows a smarter presentation of the actual state-of-the-art literature, this adds value to existing databases by providing specific aggregation of informations most ot the time isolated or even not addressed in the upstream databases.
Enrichment
Enriched functional and structural informations are primarily assembled from external reference databases. In a second time RESPIRE methods are triggered to produce sequence analysis, family annotation, 3D structures when possible, and internal cross-references for rapid database browsing. All protein entries are processed regularly.
Visualisation
In order to provide an interactive view of the current protein knowledge for a given protein, an interactive display of structures or models of RBC proteins is proposed. For a given protein of interest, the user can also map the position of known variants on the protein structure to estimate its position and evaluate the consequences of these mutations on the protein structure. For more informations on the mutation information, the user can click on the mutation to see the relevant upstream data.
Dedicated membranous annotations
Most interesting proteins in the RBC are membrane proteins: they participate in blood homeostasy but also define some RBC antigens. However, there is a paucity of experimental structures due to the difficulty of resolving their structure using commons methods (see the current list from Stephen White). To reduce this structural gap, we put a strong effort on providing models for these protein using specific membrane structure prediction methods. For some proteins, a more extensive work was performed at the DSIMB laboratory, and efforts are ongoing for producing more advanced models. In both membranous and non-membranous protein cases, we chose to offer to the user models, advertised as such, with a confidence index. This allow a preliminary indication of the protein structure, where experimental data is lacking.
Browsing the database
In order to help users in finding rapidly a specific protein, or a category of RBC proteins, there are guided entry menus, a drop-down list of proteins present in the database or a searchable text box. In many places, cross-references allow to browse the database by ontology categories, subcellular location, ... This hybrid access should address most needs.
The aquaporin entry is detailed below
Function
This tab contains the last update of the protein description from UNIPROT.

Protein expression
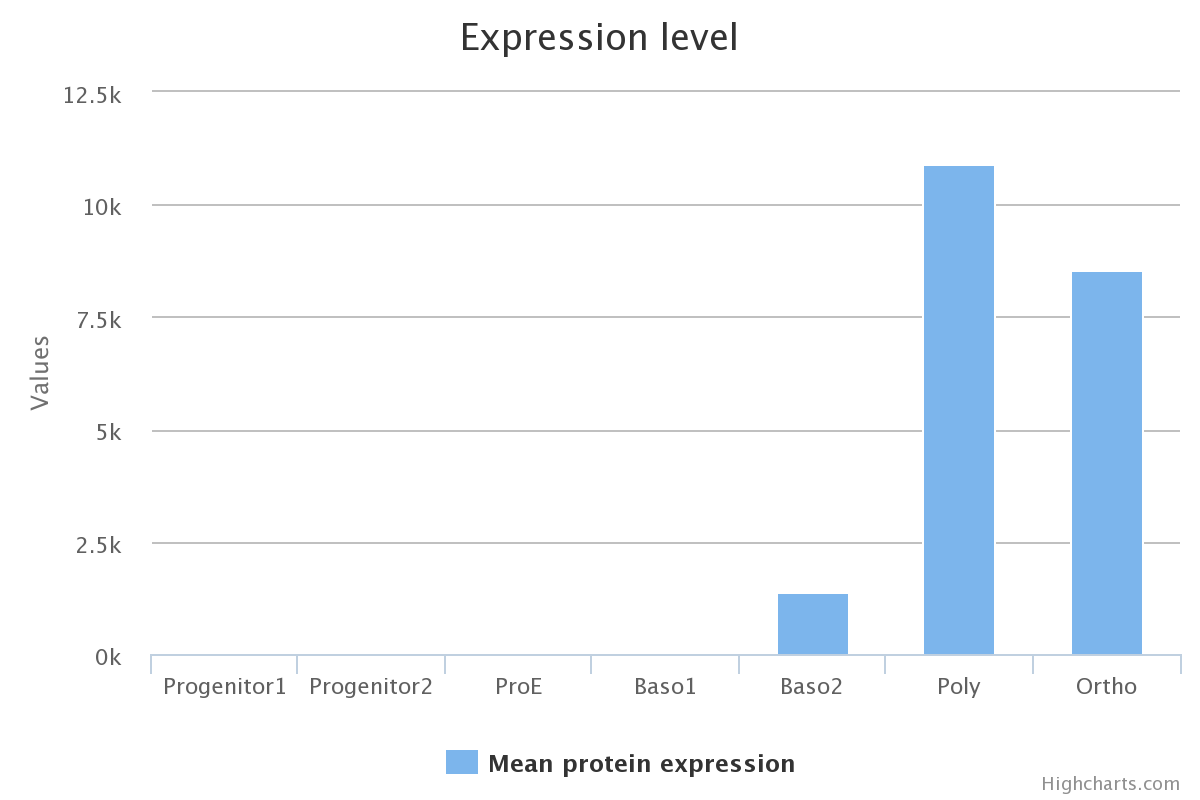
Protein expression levels were quantified using mass spectroscopy, cell differentiation stages are presented in the Erythropoiesis tab. Details of protein quantification and caracterisation is described in the original article of Gautier and co-workers.
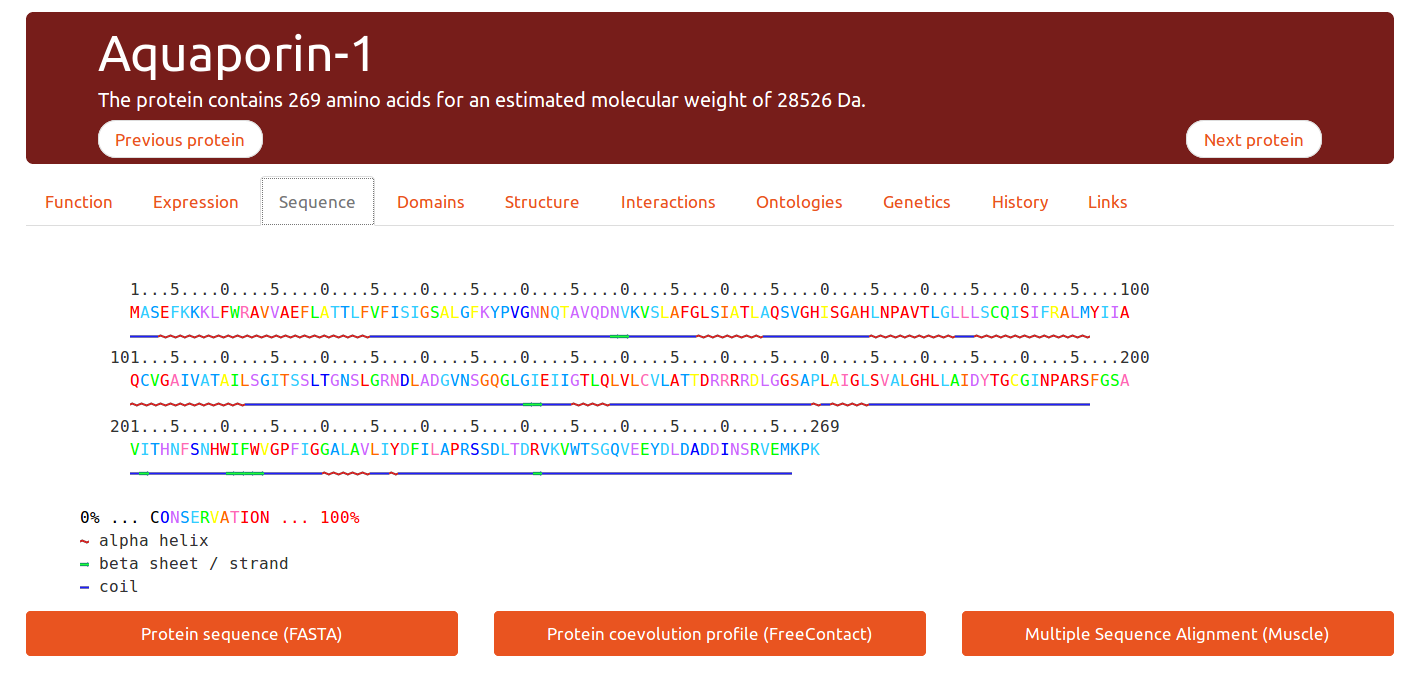
Sequence
This tab displays a pre-computed sequence conservation index per position, from no conservation (0% blue) to highly conserved (100%, red). It is also possible to download the multiple alignment used for calculating these values.
A downloadable co-evolution profile is also available to add a valuable information on protein distance contacts.
Domains
The Domains tab displays a graphical decomposition of the protein organisation into sub-domains in different databases. By overing on a domain scheme, it is possible to see its exact boundaries and/or the domain definition. When possible, links redirect to RESPIRE entries, otherwise the user is redirected to external references.
{kind=link}
Structure
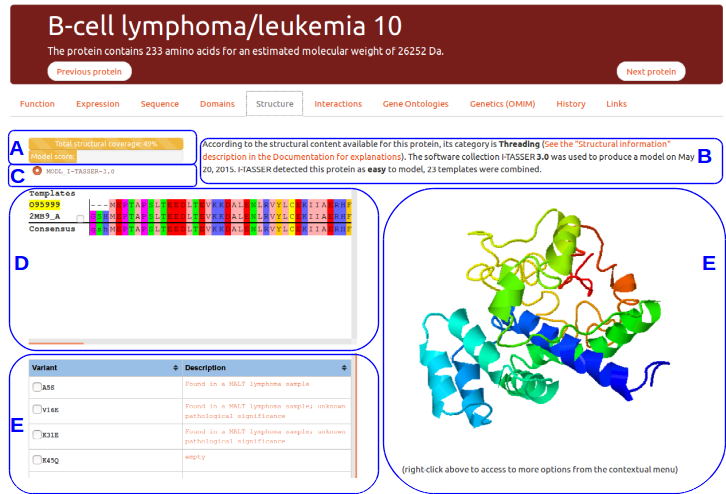
This tab gives details about the protein structural content available in the litterature and about models produced specifically for RESPIRE. This example presents the view available for B-Cell lymphomia/leucemia 10 protein.
(A) Protein structural status and structure quality evaluation
The top progress bar indicates the total protein structural coverage calculated using all the detected templates for this protein. A multiple sequence alignment of all template sequences is performed against the protein sequence. The number reported here indicates the percentage of residues for which one amino acid position can be modelled from a template.
The second progress bar indicates an evaluation of the best model quality score evaluated using the prediction method score, or experimental content if it exists. The model evaluation is only indicative, these models should not be taken as is without expert knowledge.
Both bars will display a different color according to the confidence one should have on the displayed data: red for lack of structural data, orange for putative models, violet for high quality models, and green for experimental structures.
(B) and (C) Structural templates information
This tab allows to select any template related to the protein entry. By default the best template or models is automatically loaded. The template details are provided to the user for the model when required or from the upstream structure file (PDB) when possible.
(D) Templates selection
All known templates are displayed as a list in the multiple sequence alignment window. The reference sequence is highlighted in yellow using its uniprot unique identifier.
(E) Interactive struture display
This zone allows to manipulate in three-dimension the structure of interest. A right-click on the molecule window will trigger a menu allowing more complex interactions. More actions are described in the official documentation of JSMol.
(F) Known variants and mutations
Known variants and mutations can be displayed on the protein structure when possible (all amino acids may not be available in experimental structures). PDB structures were specifically processed to ensure a correct correspondance in amino acids numbering.
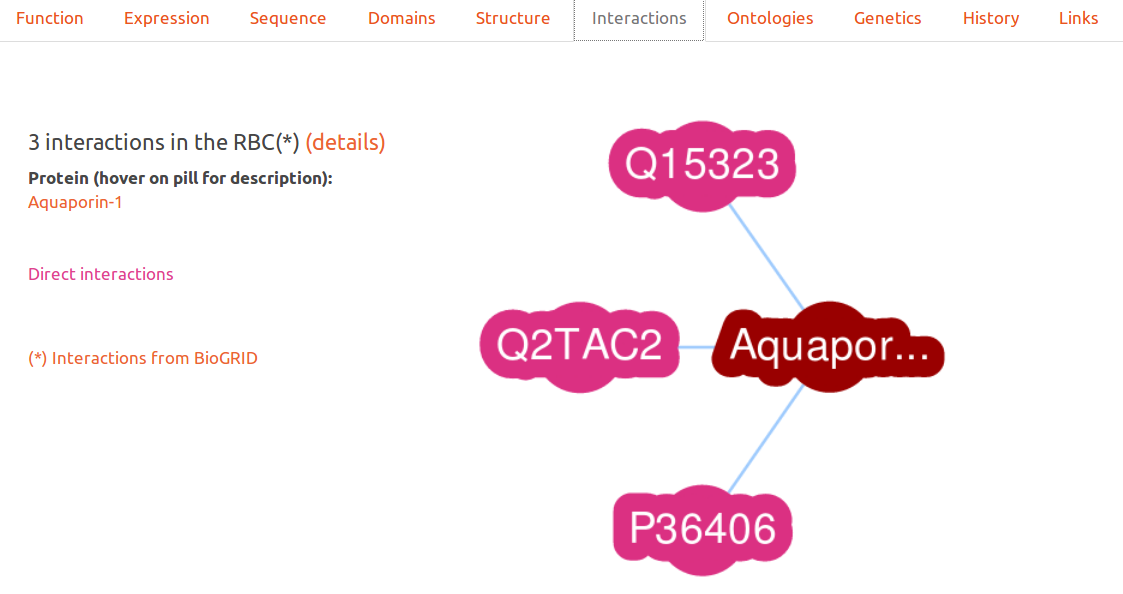
Interactions
This interactive graph displays proteins in RESPIRE which are indicated as interacting in the biogrid project. Only proteins found in the Red Blood Cell are presented to the user.
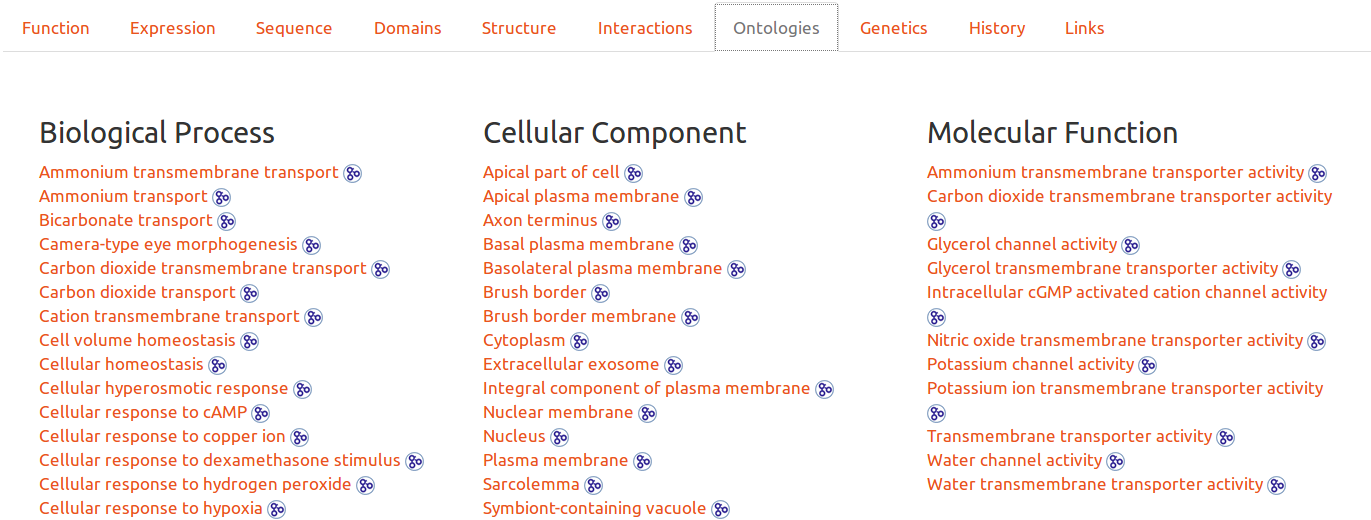
Gene Ontologies
Ontologies are another possibility to browse the database. By clicking on any entry, the user will obtain all entries related to the ontology. It is also possible to see the ontology definition by hovering ont its link, or to click on the icon to be redirected to the upstream detailed definition.
Genetics (OMIM)
In this tab, Online Mendelian Inheritance in Man entries are processed (with autorization) to show clinically relevant information. Since these entries can be of very variable lengths, we chose to present only the most representative information to the user, with a link to the upstream entry for further exploration. These upstream entry will also contain OMIM specific additional links not incorporated here for clarity.
Erythropoiesis is the biological process where immature hematopoietic cells progressively differentiate into enucleated cells.
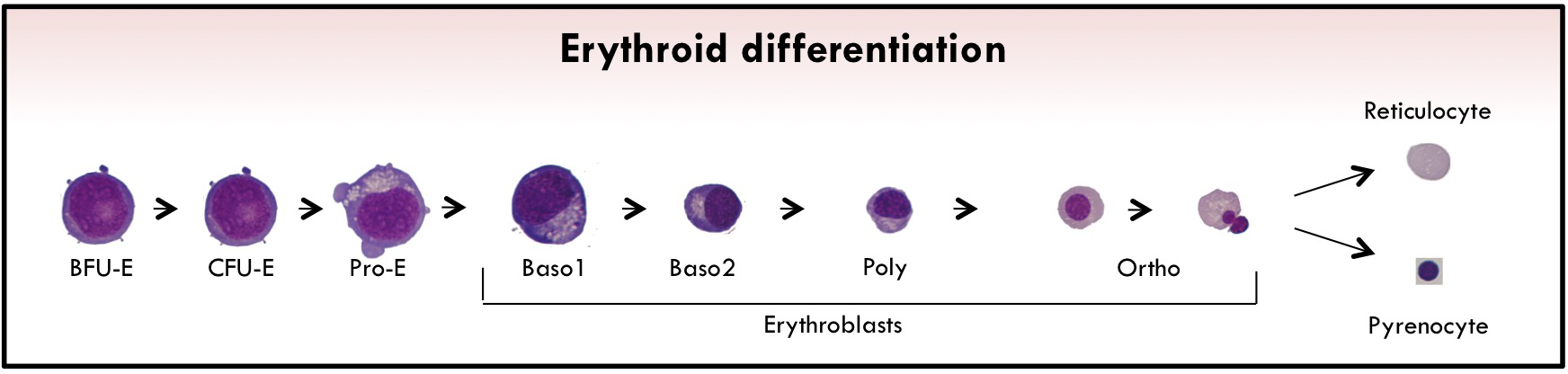
The first erythroid-committed progenitors are burst-forming units-erythroid (BFU-Es). Upon erythropoietin (EPO) stimulation, BFU-Es differentiate into late erythroid progenitors called colony-forming units-erythroid (CFU-Es).
The last phase of erythropoiesis is called terminal differentiation. In this step, several morphologically recognizable precursors are successively produced: proerythroblast (ProE) cells and basophilic I and II (Baso1 and Baso2), polychromatophilic (Poly), and orthochromatic (Ortho) erythroblasts.
During this process, the size of the cells gradually decreases, and they synthesize large amounts of hemoglobin (Hb) and reorganize their membrane together with a nuclear condensation. At the end of terminal erythroid differentiation, Ortho cells expel their nucleus, which is surrounded by plasma membrane with a small amount of cytoplasm, to generate a pyrenocyte. This cell stage is rapidly engulfed by macrophages of the erythroblastic niches.
The remaining cell is the (pre-)mature Red Blood Cell called reticulocyte which completes its maturation in the bloodstream to form erythrocyte. During this enucleation process, several proteins appear to be actively sorted between pyrenocytes and reticulocytes, although the extent of this active sorting process remains unclear.
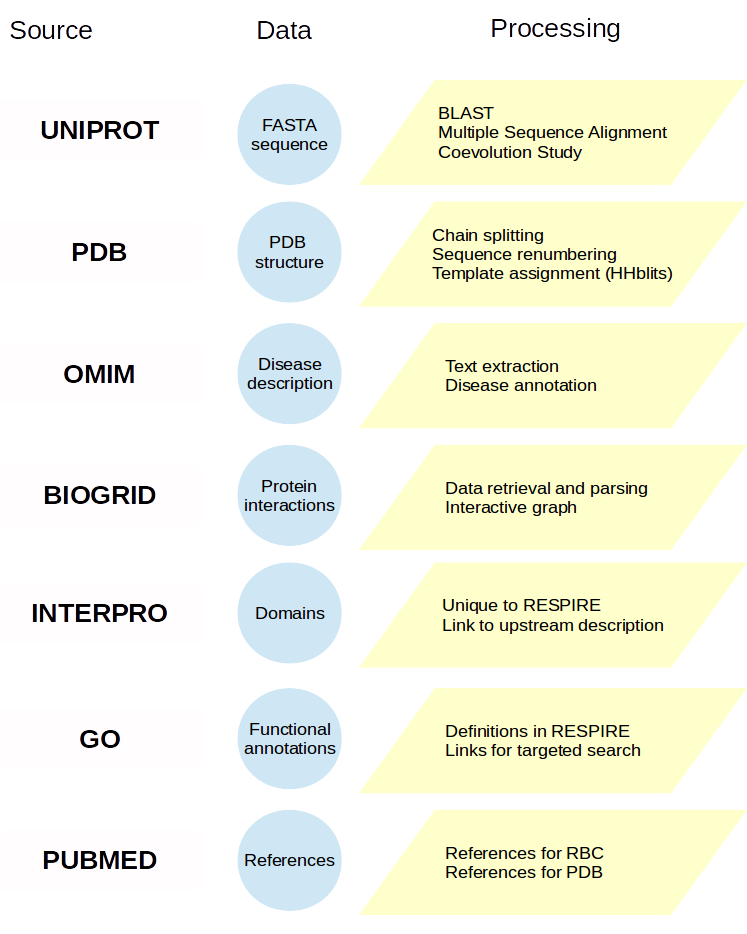
A protein entry card is primarily filled in from the UNIPROT entry to retrieve reference unique identifiers, then enriched by processing external sources or using RESPIRE methods. The diagram below indicates most reference databases and the processing performed. More details are available in the publication, but you can also contact us.
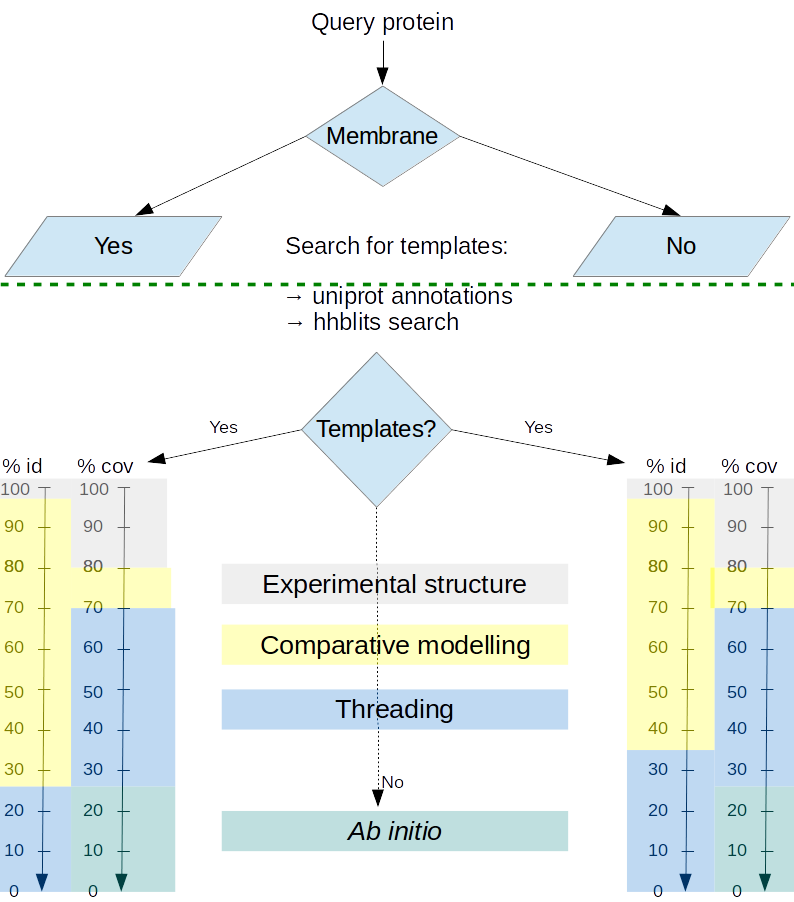
Model production
Proteins models are produced using different methods according to the difficulty of producing models from existing experimental data.- When the protein structure is experimentally available, the molecule is displayed without modification.
- When homolog structures possess a pairwise percentage of identity above 35% and it is possible to reconstruct more than 70 % of the protein of interest using these homologs as templates, comparative modeling is performed.
- When no homolog, or combination of homologs is able to satisfy these criteria, a threading method is applied using the best performing CASP threading method, I-TASSER.
- When no homolog can be detected, a less reliable prediction algorithm for ab initio approach is then applied using the ROSETTA commons tools, with a specific treatment of membrane proteins whenever applicable.
This focus on membrane proteins is particularly important since some of them have a high therapeutic interest but, on the other hand, membrane proteins are much less characterized experimentally than soluble globular proteins. The search for a better model prediction is performed weekly. If homologs can be performed, or experimental constraints allow the refinement of the model, it will be processed within another week.
The figure belows presents the database model organisation.
RESPIRE name
RESPIRE stands for Repository of Enhanced Structures of Proteins Involved in the Red Blood Cell Environment. This is also a verb in French which means "to breathe".
Can you provide a "model this for me button"?
Short answer: no. Due to the important computational cost for processing entries, it is not possible for now to allow this possibility. This situation is not desesperate, you can use other existing web services.
I would like to see feature abc added, is it possible?
Nothing is impossible, just ask and we shall see for future updates.
Some protein entries seem incomplete
First, this can be a bug, please tell us if you think this is the case. For most entries, though, there is simply not enough upstream data, or it is impossible to produce valid structural models using state-of-the-art methods for now.
The protein function seems old, isn't there any update?
The function description comes form the upstream UNIPROT entry. Although every UNIPROT card is updated regularly, most of the time many fields, including the protein function, will not be updated. We process periodically every UNIPROT entry, but the function (and the date of modification) is only updated when it changes.
Why is my protein xyz not listed in your database?
Many publications list the protein composition of Red Blood Cells. We chose to use the most extensive study from Goodman and co-workers to set up a relatively conservative initial protein content. If you think an important protein is missing in our database, just contact us. We will do our best to consider any valuable comment.
Data
Data available on this web site are available under the creative commons Attribution-NoDerivs 4.0 (CC BY-ND). This is the same policy as for the Uniprot consortium.
Data coming from external sources are processed with authorizations when required or comply to the databases policy. In case of any doubt, please use upstream database policies as reference. In case there is any doubt, just contact us.
Can I use data for my own research work?
Yes, for sure. As it is common for scientific work, please cite us. Pay attention however to proteins structures since their usage require expert knowledge.