Here, I put some of my +/- useful scripts... Hope they are useful ! Any feedback is welcome.
All these scripts/programs are intended to run under Linux on classical PC workstations, I do not provide any support or advice under other OS or platforms, thanks !
Make an index for 2 leaflets of a bilayer with GROMACS
(added Nov 2006)
Source code
Find here the script make_2leaflets_index. Rename it with a ".py" extension:
mv make_2leaflets_index make_2leaflets_index.py
Description
This script reads a pdb file containing a bilayer, and generates an input script for make_ndx in order to get an index for the lower and upper leaflets. The default atom name is "P" but you can change the variable REF_ATOM if you want to use another reference atom.
WARNING: the script assumes that the bilayer normal is aligned along Z.
Usage with a pipe:
python make_2leaflets_index.py bilayer.pdb | make_ndx -f bilayer.pdb \ -o bilayer.ndx
g_tilt: calculate the tilt angle of a transmembrane helix within GROMACS 3.3.1
(added Jun 2007)
Source code
Find here g_tilt_gmx331.c which is the source code of g_tilt for GROMACS 3.3.1.
Description
This program calculates the tilt angle between the axis of an alpha-helix and the normal to the membrane (it is thus intended for helices of membrane proteins/peptides). The helix axis is determined using the first eigen vector of the inertia matrix of the backbone atoms, while the normal to the membrane is the third eigen vector of the inertia matrix of the phosphorous atoms of one leaflet. You have to use an index file that contains 2 basic information:
- 1) the atoms that define the backbone of the helix (e.g. Calphas, or all bb atoms)
- 2) those defining one leaflet of the bilayer (e.g. P atoms for a phospholipid bilayer).
By convention, the normal to the membrane will be oriented in the same way as the helix axis, which will result in a tilt angle within the range [0-90] deg. If the option -Z is set, the Z axis will be taken as the normal instead of being calculated (I advice you to use this option since I saw funny things occuring during eigen vectors calculation of the normal). Of course it assumes that the bilayer normal is aligned along the Z axis.
Installation
Here are the steps to follow to compile g_tilt (at least they work on regular PCs under LINUX).
- cd to the gromacs directory of installation (let's call it $GMX)
cd /go/to/the/gromacs/dir
- copy g_tilt_gmx331.c to $GMX/src/tools/g_tilt.c
cp /wherever/you/put/g_tilt_gmx331.c src/tools/g_tilt.c
- edit $GMX/src/tools/Makefile.am, add 'g_tilt' in the section bin_PROGRAMS and save
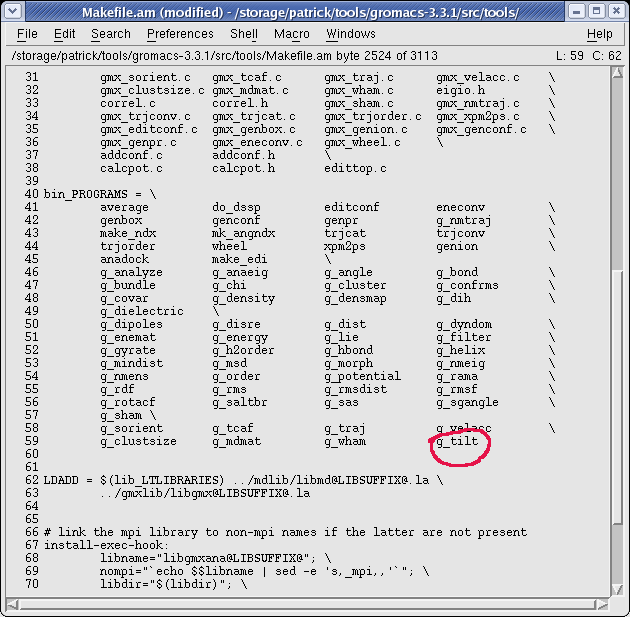
- run 'aclocal' and 'automake' in the $GMX directory.
cd /go/to/the/gromacs/dir aclocal automake
This should generate the file $GMX/src/tools/Makefile.in with all directives concerning g_tilt. - launch gromacs installation normally with ./configure (plus your options), make, make install
./configure --my-beautiful-options make make install
g_rotation: calculate the azimuthal rotation angle of a transmembrane helix within GROMACS 3.3.1
(added Jun 2007)
Source code
Find here g_rotation_gmx331.c which is the source code of g_rotation for GROMACS 3.3.1.
Description
g_rotation calculates the azimuthal rotation of an alpha-helix. This only works for helices of transmembrane proteins/peptides, that make a significant tilt and that are not completely horizontal. You have to use an index file that contains 3 basic information:
- 1) the atoms that define the backbone of the helix
- 2) those defining the upper leaflet of the bilayer
- 3) one Calpha of the residue of reference (taken as the 0 deg rotation)
This rotation is defined between the vector giving the direction of the tilt (vector orthogonal to the helix axis, and within the tilt plane [this latter is defined by the normal to the upper leaflet (approximately (0,0,1)) and the helix axis]) and a vector orthogonal to the helix axis that passes by the Calpha of the residue of reference. The positive rotation is defined in the anti-clockwise direction. This azimuthal rotation is exactly calculated as in Strandberg & al. (Biophys. J., 2004, 86, 3709-21) and Özdirekcan & al. (Biochemistry, 2005, 44, 1004-12). If the Calpha of the residue of reference coincides with the direction of the tilt, the rotation is 0 deg.
If the option -Z is set, the Z axis will be taken as the normal instead of being calculated. Of course it assumes that the bilayer normal is aligned along the Z axis. By default, g_rotation gives the azimuthal rotation within the range -180/180, or 0/360 if the option -range 360 is set.
WARNING:
- The definition of the azimuthal rotation is ambiguous when the tilt is small (e.g. close to 0 deg), you might have huge fluctuations in such a case.
- I advice you the use of the -Z option both for g_tilt and g_rotation, as I noticed funny effects during the normal (to the membrane) determination with the matrix diagolanization: sometimes it returns Nan. I did'nt have time to check this carefully (will do that when I have time :-)). A quick look shows that the normal calculated in this way undergoes angle fluctuations (compared to the Z axis) of only +/- 5 deg. Thus with the -Z option, you're safe !
Installation
Just follow exactly the same steps as for g_tilt described above.
I wrote g_tilt and g_rotation with the help of Gaëlle Debret and Suat Özdirekcan, thanks to them !
more to come...
back to top
