Principle
The protein structures are classically described as composed of two regular states, the alpha-helices and the beta-strands and one non-regular and variable state, the coil. Nonetheless, this simple definition of secondary structures hides numerous limitations. In fact, the rules for secondary structure assignments are complex. Thus, numerous assignment methods based on different criteria have emerged leading to heterogeneous and diverging results. In the same way, 3 states may over-simplify the description of protein structure; 50% of all residues, i.e., the coil, are not described even it encompass precise local protein structures.
Description of local protein structures have hence focused on the elaboration of complete sets of small prototypes or "structural alphabets", able to analyze local protein structures and to approximate every part of the protein backbone(Offmann et al., Cur Bioinf, 2007,Joseph et al 2010). The principle of a structural alphabet is simple. A set of average local protein structures is firstly designed. They approximate (efficiently) every part of the structures. As one residue is associated to one of these prototypes, the 3D information of the protein structures can be translated as a series of prototypes (letters) in 1D, as the amino acid sequence.
We have so proposed an efficient way to describe protein structures with a novel Structural Alphabet (Benros et al, 2006) and improve the prediction from the sequence using Support Vector Machines and evolutionary information (Bornot et al., 2009).
However, protein structures are not rigid macromolecules. We have so proposed to use the results from the prediction of this Structureal Alphabet to innovative propose prediction methodology (Bornot et al., 2011) not based only on the flexibility as simply the normalized B-factor, but also using results from molecular dynamics, i.e., normalized Root Mean Square Fluctuations (RMSF).
From this dual description, we have defined 3 classes of flexibility prediction: (i) rigid, (ii) intermediate and (iii) flexible. Our main goal was to have the less confusion between rigid and flexible classes. Moreover, we propose a confidence index which helps the scientist to know if the prediction is pertinent or not.
Here so we propose, prediction of :
- Normalized B-factor,
- Normalized RMSF,
- Three classes: rigid, intermediate and flexible
- Confidence index.
Local Structure Prediction Method
The prediction method relies on an expert system. For each local structural class s represented by its LSP, an expert (LSP-expert) is trained to optimally discriminate between fragment sequences associated to s (positive examples) relative to other classes (negative examples).
For a target protein sequence, PSI-BLAST searches are performed. A Position-specific Scoring Matrix (PSSM) is calculated for the complete protein sequence and is then cut into overlapping 11-residue long fragment sequences. For each fragment, a local structure prediction is performed. Each LSP-expert computes a compatibility score of a target sequence window with its class. The 120 scores are then ordered and finally, a jury selects the 5 top-scoring classes and proposes their representative LSPs as structural candidates.
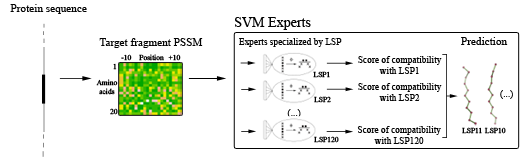
Local Structure prediction principle.
Here is an example of the results, you can get with our method :
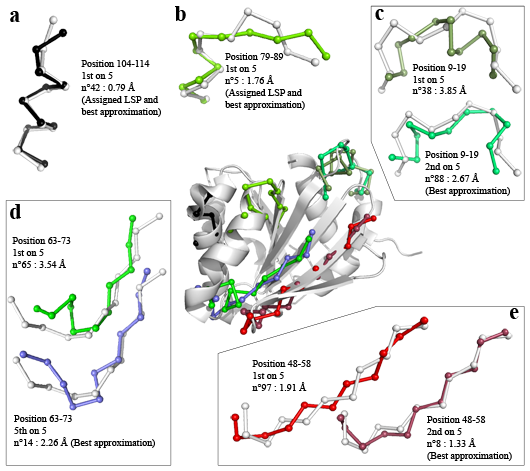
Five examples of predicted local structures for the ARL3-GDP protein (PDB code 1FZQ chain A (Hillig et al., Structure 2000), SCOP class, 175 residues).
The prediction rate obtained for this protein was 72.4 % of correct predictions. For each example, the true local structure is represented in white and the mean representative structure of the predicted structural class is in colour. These mean representatives are named Local Structure Prototypes or LSP. They are numbered from 1 to 120. Five examples are shown on the periphery of the figure and are also represented on the global structure in the centre.
The two first examples (a) and (b) refer to the structural predictions of a helical core and of a helical exit respectively. In both cases, the top-scoring candidate corresponded to the assigned LSP, i.e. lowest Calpha-RMSD (Root Mean Square Deviation) from the true local structure.
The third example (c) corresponds to a connecting local structure. The first-ranked LSP candidate was 3.85 Å Calpha-RMSD from the true local structure. The assigned LSP, numbered 14, which was 2.24 Å Calpha-RMSD from the true structural fragment, was not among the 5 candidates. Nevertheless, in the candidate list, an approximation of 2.67 Å from the true structural fragment, was obtained with LSP 88. This latter adopts a shape and an orientation of the C-terminal region similar to the true fragment.
The two last examples concern extremity of extended and extended structures (examples (d) and (e)). Regarding the extended edge structure, the top-scoring candidate LSP 64 was far from the true local structure (Calpha-RMSD value equals to 3.54 Å) but the last candidate of the list, LSP 14, achieved the best geometrical approximation, 2.26 Å Ca-RMSD, very close to the corresponding value (2.20 Å) of the assigned LSP 110. Finally, for the prediction of the extended core structure, the first ranked candidate, LSP 97, yielded a 1.91 Å Calpha-RMSD value with a good orientation of the N-terminal extremity. The next candidate in the list, LSP 8, corresponded to the best approximation (Calpha-RMSD value equalled to 1.33 Å). Among the five candidates, four of them exhibited correct approximations although the assigned LSP 96 (with 1.05 Å Calpha-RMSD) was not proposed.
Flexibility Prediction Method
The flexibility of protein local structures was studied through (i) the B-factor of X-ray experiment and (ii) the fluctuation of residues during molecular dynamics simulations. Specific properties depending on the local structural environment were enlightened.
The flexibility prediction method relies on the local structure prediction. It takes advantage from the relationships between the dynamic properties of the target sequence and those associated to the structural candidates proposed by our local structure prediction method.
Each one of the 120 structural classes was assigned to one of the 3 flexibility classes (0=rigid, 1=intermediate, 2=flexible) according to its observed dynamic properties. In the same way, it is also associated to a mean normalized B-factor and to a mean RMSF (Root mean square fluctuation, from molecular dynamic simulations).
Thus, for a target sequence, the local structure prediction is performed and proposes the 5 best candidates. From this point, the predicted flexibility class issimply obtained by calculating the rounded average of the flexibility classes of the 5 candidates. In the same way, the predicted normalized B-factor (RMSF) is obtained by calculting the average mean normalized B-factor (RMSF) of the 5 structural candidates. Hence, no training on the data was performed. The prediction reflect the informativity of structural prediction from sequence for flexibility.
References
Benros C., de Brevern A.G, Etchebest C., Hazout S. Assessing a novel approach for predicting local 3D protein structures from sequence Proteins (2006) 62(4) 865-80 [PUBMED]
Offmann B., Tyagi M., de Brevern A.G. Local Protein Structures Current Bioinformatics (2007) 2: 165-202 [PUBMED]
Bornot A., Etchebest C., de Brevern A.G. A new prediction strategy for long local protein structures using an original description Proteins (2009) 76(3):570-87 [PUBMED]
Joseph A.P., Agarwal G., Mahajan S., Gelly J.-C., Swapna L.S., Offmann B., Cadet F., Bornot A., Tyagi M., Valadié H., Schneider B., Etchebest C., Srinivasan N., de Brevern A.G. A short survey on Protein Blocks. Biophysical Reviews (2010) 2(3):137-145. [PUBMED]
Bornot A., Etchebest C., de Brevern A.G. Predicting Protein Flexibility through the Prediction of Local Structures. Proteins (2011) 79(3):839-52 [PUBMED]
de Brevern A.G., Bornot A., Etchebest C., Gelly J.-C. PredyFlexy : a protein flexibility prediction from the lone information of the sequence. in preparation
Authors
Alexandre de Brevern
Aurelie Bornot
Catherine Etchebest
Jean-Christophe Gelly