Olivia Doppelt-Azeroual PhD
Year 2009
- Doppelt-Azeroual O.
Développement dune nouvelle méthode performante de classification des surfaces protéiques dinteraction.
Optimisations et extensions du logiciel MED-SuMo.
Doctorat de l'Université Paris Diderot - Paris 7 - Spécialité : Analyses de Génomes et Modélisation Moléculaire (2009) 182 p.
Thèse en convention CIFRE avec la société MEDIT-SA.
Les succès des projets Génome sont une conséquence directe de lamélioration des techniques de séquençage à haut débit. Le nombre de séquences protéiques accessibles a ainsi augmenté de manière exponentielle, mais la quantité de séquences nayant aucune annotation fonctionnelle grandit elle aussi très rapidement. La fonction dun gène sexprime principalement au travers de son produit, la protéine, dont le rôle est directement lié à sa structure tridimensionnelle (3D). La résolution dune structure de protéine issue dun gène de fonction inconnue est un moyen dappréhender les mécanismes biochimiques. Lamélioration des méthodes de résolution de structures protéiques a permis des progrès en biologie structurale tel que lamélioration de la qualité des structures et surtout laugmentation quadratique du nombre de structures disponibles dans la Protein DataBank.
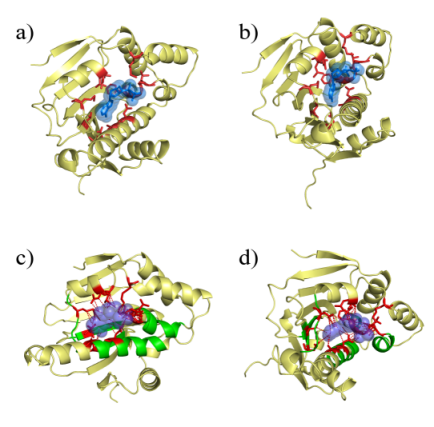 La fonction dune protéine se définit par ces interactions avec un (ou plusieurs) partenaire(s) spécifique(s). Ces interactions sont la base des mécanismes biologiques majeurs. Les interactions protéine-ligand ont des rôles cruciaux dans les fonctions de transport et de transmission de signaux cellulaires. La détection et la comparaison de surfaces dinteraction sont ainsi des étapes essentielles pour annoter fonctionnellement les protéines, et surtout dans le cadre de lélaboration de nouveaux médicaments.
La fonction dune protéine se définit par ces interactions avec un (ou plusieurs) partenaire(s) spécifique(s). Ces interactions sont la base des mécanismes biologiques majeurs. Les interactions protéine-ligand ont des rôles cruciaux dans les fonctions de transport et de transmission de signaux cellulaires. La détection et la comparaison de surfaces dinteraction sont ainsi des étapes essentielles pour annoter fonctionnellement les protéines, et surtout dans le cadre de lélaboration de nouveaux médicaments.
Le logiciel SuMo et son produit dérivé à vocation industrielle MED-SuMo représentent une des premières approches basée sur la position relative des groupements chimiques fonctionnels élémentaires disponibles à la surface des protéines, pour comparer les surfaces dinteraction. Elle possède certaines analogies avec dautres méthodes telles CavBase et SiteEngine. Toutefois, son approche est particulièrement innovante ; son originalité réside dans ses descripteurs, les « Surface Chemical Features » (SCFs) qui, rassemblés en triplets, forment un graphe. Ce dernier constitue la clé des performances élevées de lheuristique de comparaison, procédure se basant sur la détection de sous-graphes communs. Particulièrement rapide, lapproche permet la comparaison dun site de liaison à tous les sites de liaisons de la PDB en quelques minutes. La comparaison des surfaces protéiques offre un grand nombre dapplications nouvelles de drug design, basées sur lhypothèse selon laquelle les surfaces similaires interagissent avec des molécules de type similaire (Doppelt-Azeroual et al., Infectious Disorders-Drug Targets, 2009).
Mon travail de thèse repose sur trois points:
Tout dabord, jai pu participer activement à loptimisation du logiciel MED-SuMo, notamment dans le cadre de la mise en place des communications avec la nouvelle interface graphique dédiée, MED-SuMo GUI. Jai également mis en évidence lintérêt de MED-SuMo pour lannotation fonctionnelle de protéines hypothétiques aux travers de deux applications. Pour la protéine TM1012, alors que les approches classiques détudes des protéines ne donnent aucun résultat exploitable, MED-SuMo propose des fonctions biochimiques en détectant des surfaces dinteraction similaires à dautres sites de liaison dont la fonction est définie et les mécanismes daction connus (Doppelt et al, Bioinformation, 2007). Pour la protéine YBL036C, annotée comme hypothétique, la situation est différente. PSI-BLAST détecte des similitudes avec des alanines racémases ayant un taux didentité de séquence proche de 40%. ProFunc, grâce à la méthode de comparaison DALI, détecte également des similitudes avec des structures dalanines racémases. MED-SuMo confirme ici de manière précise ce que dautres approches détectent en localisant quatre alanines racémases dont les sites de liaison se superposent parfaitement avec celui de la requête (Doppelt-Azeroual et al., Infectious Disorders-Drug Targets, 2009).
En second lieu, la partie essentielle de mon travail de recherche a été le développement et limplémentation dune nouvelle méthode de classification des sites de liaison protéique, MED-SMA. Cette approche se base sur la comparaison deux à deux de tous les sites de liaison dun jeu de protéines dans le but dy extraire des régions similaires détectées non plus entre paires de sites mais pour un ensemble de sites. Deux applications ont été réalisées sur des ensembles de sites de liaison connus. Pour chacune delle, MED-SMA rassemble dans les mêmes groupes des sites détectés pour fixer des types équivalents de molécules. Une étude de la famille SCOP comprenant les HSP90; le repliement Bergerat, a été effectuée avec MED-SMA (Doppelt-Azeroual et al., Drug Design, Development and Therapy, 2009) ainsi que sur lensemble des sites du purinôme (Doppelt-Azeroual et al, soumis). Par exemple, les protéines kinase CDK2 et Aurora-A sont issues de familles différentes (CMGC et PTK) mais sont toutefois inhibées par le même type de molécules. MED-SMA détecte les similitudes de modes de liaison et regroupe leur site de liaison.
Enfin, jai participé au développement dune nouvelle méthode de conception de molécules actives de novo combinant la détection MED-SuMo de similitudes locales des surfaces des protéines avec une approche fragmentale (Moriaud et al., Journal of Chemical Information and Modeling, 2009; Oguievetskaia et al., soumis).
Indications
Soutenance le Lundi 30 Mars 2009 à 14 heures
à l'Institut National de Transfusion Sanguine, 6, rue Alexandre Cabanel 75015 PARIS (Mo Cambronne)
Jury composé de :
- Pr Gilbert Deléage, Professeur, Université Claude Bernard, Lyon 1, Rapporteur
- Dr Anne Imberty, Directeur de Recherche, CNRS, Grenoble, Rapporteur
- Pr Alessandra Carbone, Professeur, Université Pierre et Marie Curie, Paris 6, Examinateur
- Dr Cyril Daveu, Modélisateur, SANOFI-AVENTIS, Examinateur
- Pr Fernando Rodrigues-Lima, Professeur, Université Paris-Diderot - Paris 7, Examinateur
- Dr Alexandre de Brevern, Chargé de Recherche, INSERM, Directeur de thèse
- Mr François Delfaud, PDG de MEDIT-SA
Publications
-

 Doppelt O., Moriaud F., Bornot A., de Brevern A.G.
Doppelt O., Moriaud F., Bornot A., de Brevern A.G.
Functional annotation strategy for protein structures
Bioinformation (2007) Mar 19;1(9):357-9.
- Doppelt-Azeroual O., Moriaud F., Delfaud F., de Brevern A.G.
Analysis of HSP90 related folds with MED-SuMo classification approach
Drug Design, Development and Therapy (2009) 3:5972.
- Moriaud F., Doppelt-Azeroual O., Martin L., Oguievetskaia K., Koch K., Vorotyntsev A., Adcock S.A., Delfaud F.
Computational Fragment-Based Approach at PDB Scale by Protein Local Similarity.
J Chem Inf Model. (2009) 49(2):280-94.
- Doppelt-Azeroual O., Adcock S.A., Moriaud F., Delfaud F.
MED-SuMo Applications.
Infectious Diseases - Drug Targets. (2009) 9(3):344-57.
-
Oguievetskaia K., Martin-Chanas L., Vorotyntsev A., Doppelt-Azeroual O., Brotel X., Adcock S.A., de Brevern A.G., Delfaud F., Moriaud F.
Computational Fragment-Based Drug Design to explore the hydrophobic sub-pocket of the Mitotic Kinesin Eg5 allosteric binding site
Journal of Computer-Aided Molecular Design (2009) 23 (8):571-82.
-
Doppelt-Azeroual O., Delfaud F., Moriaud F., de Brevern A.G.,
Classification of binding sites with MED-SuMo Multi approach: an application on Purinome
Protein Science (2010) 19(4):847-67.
Alexandre G. de Brevern
Last Modification : March 2024
Last Modification : March 2024