Alexandre de Brevern's website
|
adress :
INSERM UMR_S 1134, DSIMB, Univ. Paris Diderot, Sorbonne Paris Cite INTS, lab. of excellence GR-Ex 6, rue Alexandre Cabanel 75739 Paris Cedex 15, France |

|
DSIMB

I'm a bioinformatician researcher at the French National Institute of Health and Medical Research (INSERM) since 2002. I'm currently a Senior Researcher working in a Bioinformatics lab named DSIMB (Dynamics of Structures and Interactions of Macromolecules in Biology). Part of our lab is located at the University of Reunion Island.
The unit is a mixed unit between INSERM and University Paris Diderot, a University of Sorbonne Paris Cite. We are located at the French National Institute of Institute of Blood Transfusion (INTS) and are also part of Laboratory of Excellence GR-Ex. We are the second team of INSERM UMR_S 1134 (Integrated biology of Red Blood Cell) unit directed by Yves Colin.
DSIMB is a Bioinformatics Team devoted to different thematics (molecular modelling, molecular dynamics, protein docking, ...). Its particularity is to (i) develop novel bioinformatics approach and (ii) have biomedical applications using both classical approaches and our novel developments. This research unit (so called EBGM) has been created and directed by Pr. Serge Hazout until 2005, the current director is Pr. Catherine Etchebest.
This part is slightly outdated:.
local sequence - structure relationships
structural alphabets: The protein structures are classically described as composed of two regular states, the alpha-helices and the beta-strands and one non-regular and variable state, the coil. Nonetheless, this simple definition of secondary structures hides numerous limitations. In fact, the rules for secondary structure assignments are complex. Thus, numerous assignment methods based on different criteria have emerged leading to heterogeneous and diverging results. In the same way, 3 states may over-simplify the description of protein structure; 50% of all residues, i.e., the coil, are not described even it encompass precise local protein structures.Description of local protein structures have hence focused on the elaboration of complete sets of small prototypes or "structural alphabets", able to analyse local protein structures and to approximate every part of the protein backbone (Offmann et al., Cur Bioinf, 2007). The principle of a structural alphabet is simple (see Figure 1a). A set of average local protein structures is firstly designed. They approximate (efficiently) every part of the structures. As one residue is associated to one of these prototypes, we can translate the 3D information of the protein structures as a series of prototypes (letters) in 1D, as the amino acid sequence.
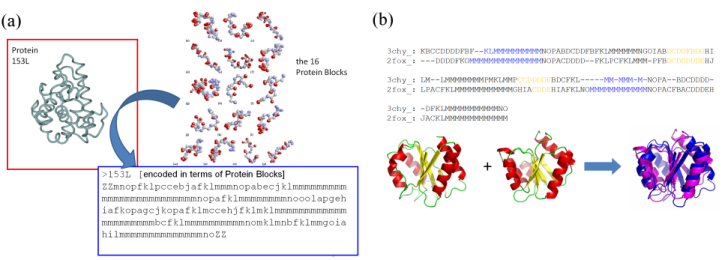
Figure 1: (a) Principle of the coding of protein structures (3D) into a series of letters (1D); (b) use of the Protein Blocks to superimpose protein structures.
The Protein Blocks: Structural alphabets have also been used to predict the protein backbone conformation and in ab initio / de novo methods. Our structural alphabet is composed of 16 mean protein fragments of 5 residues in length, called Protein Blocks (PBs, de Brevern et al., Proteins, 2000). They have been used both to describe the 3D protein backbones and to perform a local structure prediction (Proteins 2005). Our works on PBs have proven their efficiencies in the description and the prediction of long fragments and short loops (Protein Science 2002, BMC Bioinformatics 2004), to define a reduced amino acid alphabet dedicated to mutation design (European Biophysics Journal, 2007) or in the building of a transmembrane protein (Biochem Biophys Acta, 2005). We have also used this approach to compare / superimpose protein structures (see Figure 1b). For this purpose, we have used classical sequence alignment (with a dedicated substitution matrix). The assessment of this simple approach done on the classical benchmark sets was surprisingly excellent (Proteins 2006, 2008).
Researches performed in other laboratories on PBs have focussed on the building of globular protein structures, the design of peptides or the definition of binding site signatures. The features of this alphabet have been compared by Karchin and co-workers with those of 8 other structural alphabets showing clearly that our PB alphabet is highly informative, with the best predictive ability of those tested. It is the most widely used structural alphabet in the world.
For a review on PBs see Joseph et al., Biophys Rev, 2010.
This research had begun by Pr. Serge Hazout during my PhD. Most of the works are done with Pr. C. Etchebest and Dr. B. Offmann. We have a strong collaboration with Pr. N. Srinivasan (IISc Bangalore India) especially for the protein structure superimposition. It was also the core of C. Benros, M. Tyagi and Agnel P. Joseph's Ph.Ds.
HPM and the protein flexibility
Hybrid Protein Model: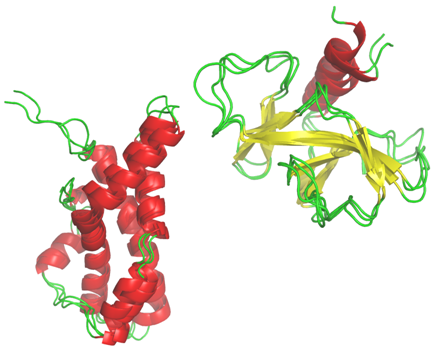 I've also developed a particular classification approach which take account of the characteristic of the protein structures. This method is related to the Self-Organizing Maps developped by T. Kohonen. It is the Hybrid Protein which allows to compact a structural databank into a limited number of clusters (Bioinformatics, 2003; Proteins, 2006, 2009, J Theo Biol, 2009).
It was the work of C. Benros during her PhD.
I've also developed a particular classification approach which take account of the characteristic of the protein structures. This method is related to the Self-Organizing Maps developped by T. Kohonen. It is the Hybrid Protein which allows to compact a structural databank into a limited number of clusters (Bioinformatics, 2003; Proteins, 2006, 2009, J Theo Biol, 2009).
It was the work of C. Benros during her PhD.
Local Structure Protein Flexibility: Protein dynamics is at the core of their biological functions. Catalysis phenomena, molecular recognition, signalling processes or protein folding are some of the essential biological events in which protein flexibility properties have a critical role. Therefore, toward the understanding of protein function at the molecular level, structural analysis need to take into account flexibility information.
The flexibility of the protein plays a crucial role in many crucial biological processes. Flexible protein regions are often not unstructured regions. By using the description of long protein fragments, we analyze the characteristics of these local protein structures. We develop a predictive approach that combines experimental data and molecular dynamic simulations. This work was done by A. Bornot for her PhD.
Protein structures
Protein Units: Protein structures can be seen as composed of single or multiple functional domains that can fold and function independently. Dividing a protein into domains is useful for more accurate structure and function determination. The commonly used principle for automatic domain parsing is that interdomain interaction under a correct domain assignment is weaker than the intradomain interaction. Some authors have proposed different methods to hierarchically split proteins into compact units smaller than protein domains, e.g. DIAL approach.We have likewise developed a method called Protein Peeling (see our website). This algorithm dissects a protein into Protein Units (PUs). A PU is a compact sub-region of the 3D structure corresponding to one sequence fragment. The basic principle is that each PU must have a high number of intra-PU contacts, and, a low number of inter-PU contacts. Protein Peeling works from the Calpha-contact matrix translated into contact probabilities. Organization of protein structures can be considered in a hierarchical manner: secondary structures are the smallest elements, and, Protein Units are intermediate elements leading to structural domains. This work is done mainly by J.-C. Gelly.
Protein functions:
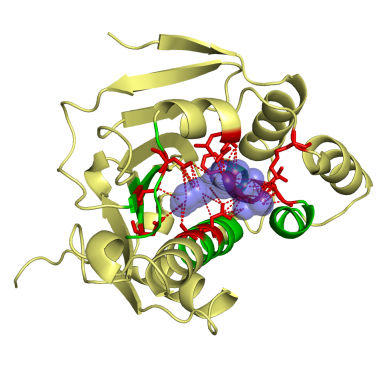 This work was a close collaboration with start-up MEDIT-SA. Ligand-protein interactions are essential at biological processes. MED-SuMo is a powerful tool to localize similar local regions associated to a defined function. It is based on a representation of macromolecules using chemical groups that are associated to heterogeneous geometrical properties. We used this approach to classify of several protein families. The gathering of binding sites with similar binding modes can be used in target based drug design applications or to predict cross-reactivity and potential toxic side effects (Bioinformation, 2007, DDDT 2009). This work was done by O. Doppelt-Azeroual for her PhD.
This work was a close collaboration with start-up MEDIT-SA. Ligand-protein interactions are essential at biological processes. MED-SuMo is a powerful tool to localize similar local regions associated to a defined function. It is based on a representation of macromolecules using chemical groups that are associated to heterogeneous geometrical properties. We used this approach to classify of several protein families. The gathering of binding sites with similar binding modes can be used in target based drug design applications or to predict cross-reactivity and potential toxic side effects (Bioinformation, 2007, DDDT 2009). This work was done by O. Doppelt-Azeroual for her PhD.
Protein Contacts: In a recent work, we studied contacts within protein structures according to various criteria (lengths of proteins, SCOP classes, secondary structures, amino acid frequencies, accessibility). We showed that the distribution of the average contact number was clearly dependant to atoms taken as references. One of the most interesting results was the fact that contacts taken into account according to a given type of distance is not compulsorily taken into account by another one, e.g., only 22% of the observed contacts considering side-chains are found if only alpha carbons are considered. Specificities were found according to the distance in the sequence between residues in contact and some differences were observed compared to the literature. Moreover, we highlighted biases of the side-chain replacement methods. This work had been done by G. Faure and A. Bornot.
Transmembrane proteins
Methodological questions: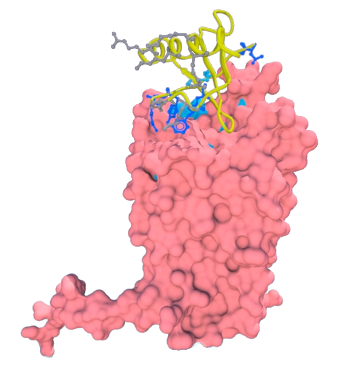 Transmembrane protein represents about ~25% of proteins coded by genomes. They are composed of two major classes: all-alpha, e.g. rhodopsin and all-beta, e.g., Outer Membrane Proteins. They are the support of essential biological functions acting as receptors, transporters or channels. Moreover, 2/3 of the marketed drugs targets a transmembrane protein and 50% specifically a GPCR, obtaining atomic structures becomes a major axis of research. Yet, these proteins are embedded in a lipid membrane that constitutes a very specific environment. Because the proteins are strongly destabilized when extracted from their natural medium, 3D transmembrane structures are difficult to obtain experimentally. On that account, the total number of transmembrane proteins in the Protein DataBank is limited, representing ~1% of all the available structures. Thus alternative approaches are required to obtain structural information. Consequently methods aiming at constructing 3D structural models are become an important research fields for understanding biological mechanisms and interactions.
Transmembrane protein represents about ~25% of proteins coded by genomes. They are composed of two major classes: all-alpha, e.g. rhodopsin and all-beta, e.g., Outer Membrane Proteins. They are the support of essential biological functions acting as receptors, transporters or channels. Moreover, 2/3 of the marketed drugs targets a transmembrane protein and 50% specifically a GPCR, obtaining atomic structures becomes a major axis of research. Yet, these proteins are embedded in a lipid membrane that constitutes a very specific environment. Because the proteins are strongly destabilized when extracted from their natural medium, 3D transmembrane structures are difficult to obtain experimentally. On that account, the total number of transmembrane proteins in the Protein DataBank is limited, representing ~1% of all the available structures. Thus alternative approaches are required to obtain structural information. Consequently methods aiming at constructing 3D structural models are become an important research fields for understanding biological mechanisms and interactions. We so developed pertinent approach dedicated to these kinds of proteins.
DARC: My work focussed on the transmembrane proteins which are implicated into numerous diseases and especially malaria. The Duffy Antigen/Receptor for Chemokine (DARC) is a seven segment transmembrane protein. It was firstly discovered as a blood group antigen and was the first specific gene locus assigned to a specific autosome in man. It became more famous as an erythrocyte receptor for malaria parasites (Plasmodium vivax and Plasmodium knowlesi), and finally for chemokines. DARC is an unorthodox chemokine receptor as (i) it binds chemokines of both CC and CXC classes and (ii) it lacks the Asp-Arg-Tyr consensus motif in its second cytoplasmic loop hence cannot couple to G proteins and activate their signaling pathways. DARC had also been associated to cancer progression, numerous inflammatory diseases, and possibly to AIDS.
We have built structural models of DARCThe chosen structural models encompass most of the biochemical data known to date (Biochem Biohys Acta, 2005). We performed protein protein docking between DARC structural models and CXCL-8 structures. We proposed a hierarchal search based on separated rigid and flexible docking.
Five main publications (methods):
- de Brevern A.G., Etchebest C. and Hazout S. (2000), Bayesian probabilistic approach for prediction backbone structures in terms of protein blocks, Proteins, 41, 271-287 [abstract]
- Etchebest C., Benros C., Hazout S. and de Brevern A.G. (2005), A structural alphabet for local protein structures: improved prediction methods, Proteins, 59, 810-827.
- Gelly J.-C. and de Brevern A.G. (2011), Protein Peeling 3D: New tools for analyzing protein structures, Bioinformatics,(2011) 27(1):132-3.
Five main publications (applications):
- Arnaud L., Saison C., Helias V., Lucien N., Steschenko D., Giarratana M.-C., Claude Prehu C., Foliguet B., Montout L., de Brevern A.G., Francina A., Ripoche P., Fenneteau O., Da Costa L., Peyrard T., Coghlan G., Illum N., Birgens H., Tamary H., Iolascon A., Delaunay J., Tchernia G., Cartron J.-P. (2010), A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia, The American Journal of Human Genetics (2010) 87(5):721-7 [abstract].
Reviews:
- Joseph A.P., Agarwal G., Mahajan S., Gelly J.-C., Swapna L.S., Offmann B., Cadet F., Bornot A., Tyagi M., Valadié H., Schneider B., Etchebest C., Srinivasan N. and de Brevern A.G. (2011), A short survey on Protein Blocks, Biophysical Reviews, 2(3):137-145 [abstract].
Alexandre G. de Brevern
Last Modification : August 2017
Last Modification : August 2017