PredyFlexy software 1.3.1
Introduction
Proteins are major components of all living cells. They are composed of a succession of amino acids.
Protein structures are mainly obtained by X-ray or Nuclear Magnetic Resonance (NMR) experiments (see Fig. 1a to 1d) which are listed in the Protein Databank (PDB, http://www.rcsb.org/). Protein sequences are classically considered as containing the whole information for their three-dimensional (3D) structure. Most proteins can exhibit global or local adaptations to perform their function.
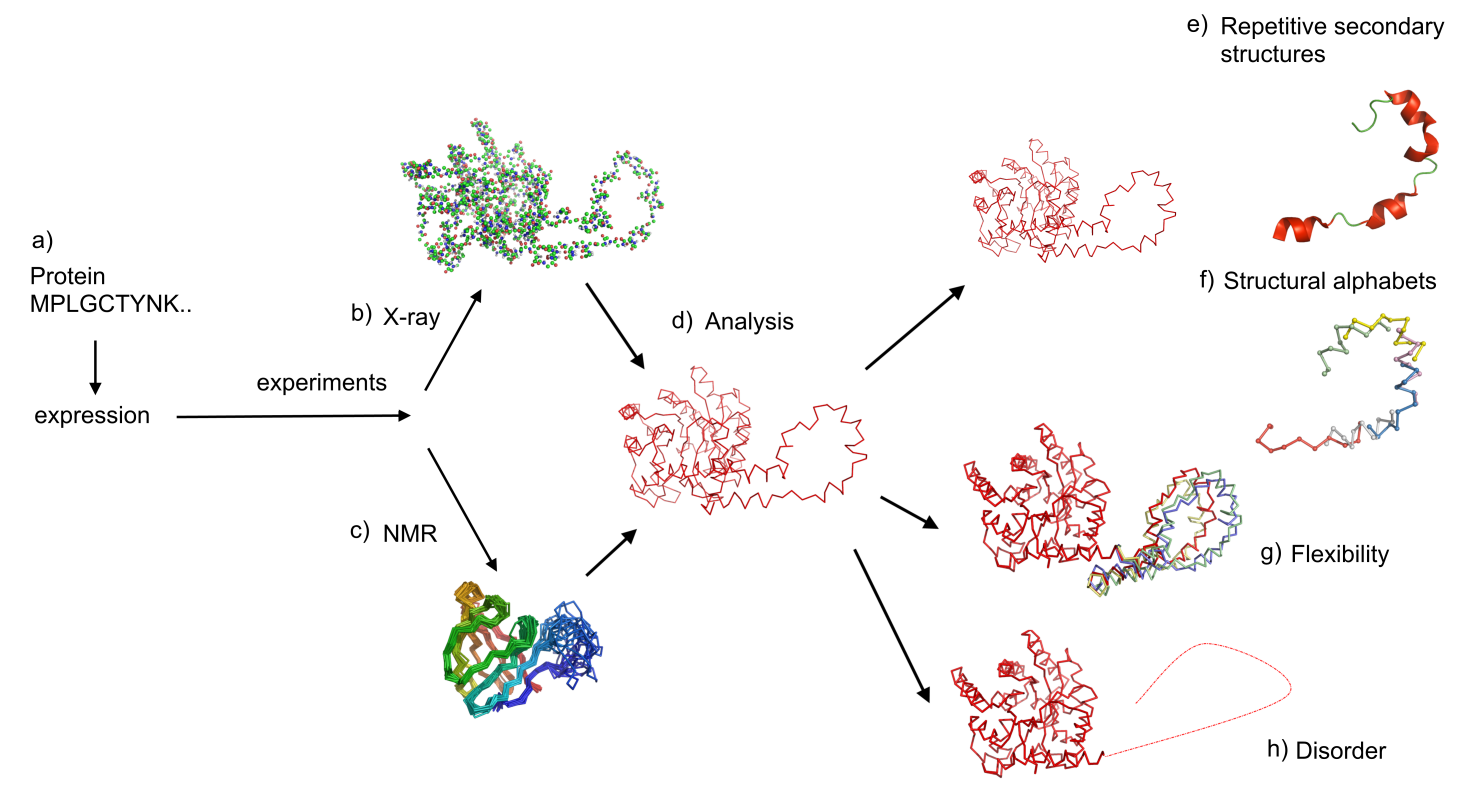
Two experimental methods measure this flexibility (see Fig. 1f) in precise regions of protein structures. The atomic mean-square displacement, or B-factor, measured during crystallographic experiments reflects an uncertainty about the position of atoms and represents the combined effects of the thermal vibrations and the static disorder. However, B-factor is not an absolute quantification of flexibility: it depends on the resolution of the structure, on the refinement procedure, on contacts in the crystal or on the structural environment. Flexibility is also indirectly highlighted by NMR experiments that show, in some circumstances, different local conformations that could correspond to different states of protein structures. The distinction between flexible, highly flexible and unstructured or disordered regions remains a difficult task.
In order to describe flexibility in a relevant way to make possible mechanical and functional analyses, we cross results from X-ray and Molecular Dynamics experiments.
Analyzing these data requires a fine knowledge and precise description of the available protein folds. Protein folds are often described as a succession of secondary structures (see Fig. 1e). Their repetitive parts (alpha-helices and helices-strands) have been intensively analyzed. Nonetheless, this description does not precisely describe the protein structures, because it fails to describe the relative orientation of connecting regions. Indeed, the coil state covering almost 50% of all residues corresponds to a large set of distinct local protein structures (see Fig. 1f). Thus, to circumvent these difficulties, other approaches were developed. They led to a new approach towards description of 3D protein structures which are now viewed as a combination of small local structures or fragments, also called structural alpahbet [1, 2, 3]. For that purpose, we have developed a library of 120 structural prototypes of 11-residue long encompassing all known local protein structures with a good approximation and specially designed for prediction from sequence [4, 5]. One of its advantages is to take into account long range interactions thanks to the significant length of the prototypes.
 Using our library, we successfully developed a local structure prediction method from sequence relying on the deduced sequence-structure relationships. We also addressed the question of the structural predictability of a sequence and defined indexes aiming at quantifying the sequence informativity on its structure [ 6].
Using our library, we successfully developed a local structure prediction method from sequence relying on the deduced sequence-structure relationships. We also addressed the question of the structural predictability of a sequence and defined indexes aiming at quantifying the sequence informativity on its structure [ 6].
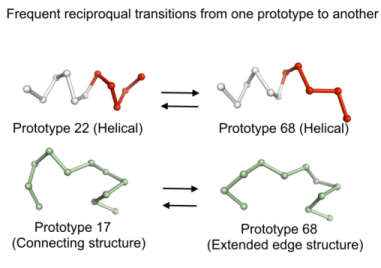
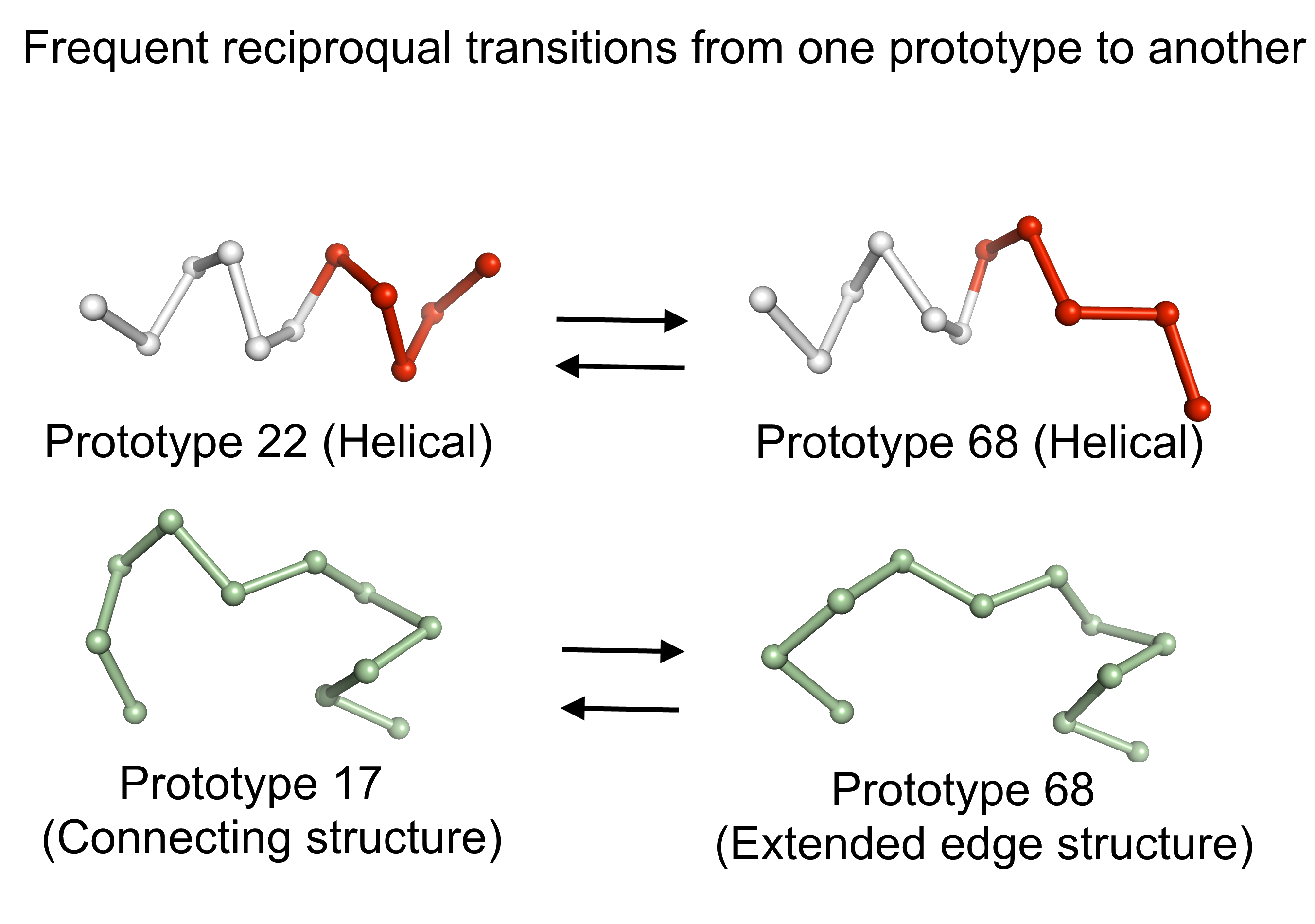
We take advantage of this fine description to observe preferential local deformations of structures (see Fig. 2). In fact, the prototypes describing a given protein structure can be changed during MD simulations, reflecting protein plasticity [7, 8]. Exploration of the conformational space is thus defined as the number of prototypes visited. Successive deformations through time and the propagation of these changes through structures are studied.
We extend now our analysis for deciphering from a sequence the putative flexible zones of a structure and the dynamics of the fragments composing it. For instance, is informativity of the sequence in relation with its structural plasticity? Relying on the integration of multiple views of flexibility, sophisticated analyses are performed for defining new indexes relating sequence to dynamics. These researches have provide new interesting tracks for predicting flexible regions and putative alternative conformers from sequence [8, 9, 10].
We have decided to provide the tool for local usage (with the most simple "usage" we hope) [10] as we have received an important number of corresponding requests from scientists and students to install it and use it locally. Indeed, the execution time is important due to the use of 120 SVMs. If you own on a big cluster it is quite faster.
In order to describe flexibility in a relevant way to make possible mechanical and functional analyses, we cross results from X-ray and Molecular Dynamics experiments.
Analyzing these data requires a fine knowledge and precise description of the available protein folds. Protein folds are often described as a succession of secondary structures (see Fig. 1e). Their repetitive parts (alpha-helices and helices-strands) have been intensively analyzed. Nonetheless, this description does not precisely describe the protein structures, because it fails to describe the relative orientation of connecting regions. Indeed, the coil state covering almost 50% of all residues corresponds to a large set of distinct local protein structures (see Fig. 1f). Thus, to circumvent these difficulties, other approaches were developed. They led to a new approach towards description of 3D protein structures which are now viewed as a combination of small local structures or fragments, also called structural alpahbet [1, 2, 3]. For that purpose, we have developed a library of 120 structural prototypes of 11-residue long encompassing all known local protein structures with a good approximation and specially designed for prediction from sequence [4, 5]. One of its advantages is to take into account long range interactions thanks to the significant length of the prototypes.
 Using our library, we successfully developed a local structure prediction method from sequence relying on the deduced sequence-structure relationships. We also addressed the question of the structural predictability of a sequence and defined indexes aiming at quantifying the sequence informativity on its structure [ 6].
Using our library, we successfully developed a local structure prediction method from sequence relying on the deduced sequence-structure relationships. We also addressed the question of the structural predictability of a sequence and defined indexes aiming at quantifying the sequence informativity on its structure [ 6].
We take advantage of this fine description to observe preferential local deformations of structures (see Fig. 2). In fact, the prototypes describing a given protein structure can be changed during MD simulations, reflecting protein plasticity [7, 8]. Exploration of the conformational space is thus defined as the number of prototypes visited. Successive deformations through time and the propagation of these changes through structures are studied.
We extend now our analysis for deciphering from a sequence the putative flexible zones of a structure and the dynamics of the fragments composing it. For instance, is informativity of the sequence in relation with its structural plasticity? Relying on the integration of multiple views of flexibility, sophisticated analyses are performed for defining new indexes relating sequence to dynamics. These researches have provide new interesting tracks for predicting flexible regions and putative alternative conformers from sequence [8, 9, 10].
We have decided to provide the tool for local usage (with the most simple "usage" we hope) [10] as we have received an important number of corresponding requests from scientists and students to install it and use it locally. Indeed, the execution time is important due to the use of 120 SVMs. If you own on a big cluster it is quite faster.
References
-
 de Brevern A.G. , Etchebest C., Hazout S.
de Brevern A.G. , Etchebest C., Hazout S.
Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks
Proteins (2000) 41, 271-287
-
Offmann B., Tyagi M., de Brevern A.G.
Local Protein Structures
Current Bioinformatics (2007) 2: 165-202.
-
Joseph A.P., Agarwal G., Mahajan S., Gelly J.-C., Swapna L.S., Offmann B., Cadet F., Bornot A., Tyagi M., Valadié H., Schneider B., Etchebest C., Srinivasan N., de Brevern A.G.
A short survey on Protein Blocks.
Biophysical Reviews (2010) 2(3):137-145.
- de Brevern A.G., Hazout S.
'Hybrid Protein Model' for optimally defining 3D protein structure fragments
Bioinformatics (2003) 19, 345-353
-
Benros C., de Brevern A.G, Etchebest C., Hazout S.
Assessing a novel approach for predicting local 3D protein structures from sequence
Proteins (2006) 62(4) 865-80
- Bornot A., Etchebest C., de Brevern A.G.
A new prediction strategy for long local protein structures using an original description
Proteins (2009) 76(3):570-87.
-
Bornot A., Offmann B., de Brevern A.G.
How flexible protein structures are ? New questions on the protein structure plasticity
Bioforum Europe (2007) 11, 24-25
-
Bornot A., Etchebest C., de Brevern A.G.
Predicting Protein Flexibility through the Prediction of Local Structures.
Proteins (2011)79(3):839-52.
-
de Brevern A.G.*, Bornot A.*, Craveur P., Etchebest C., Gelly J.-C.
PredyFlexy: Flexibility and Local Structure prediction from sequence
Nucleic Acid Res (2012) 40:W317-22.
*: authors contribute equally.
-
Narwani T.J.*.,Etchebest C.*, Craveur P., Leonard S., Rebehmed J., Bornot A., Gelly J.-C., de Brevern A.G.
in silico prediction of protein flexibility with local structure approach
submitted (2016) .
*: authors contribute equally.
Alexandre G. de Brevern
Last Modification : July 2016
Last Modification : July 2016