| The
KNOTTIN database |
|
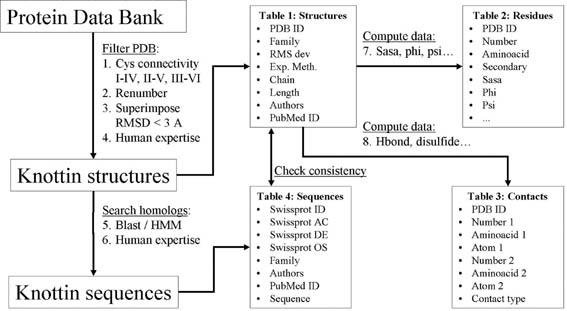
As of September 2007, the KNOTTIN database is updated using mostly
the automatic procedures KNOTER3D and KNOTER1D, according to the flow
chart shown below.
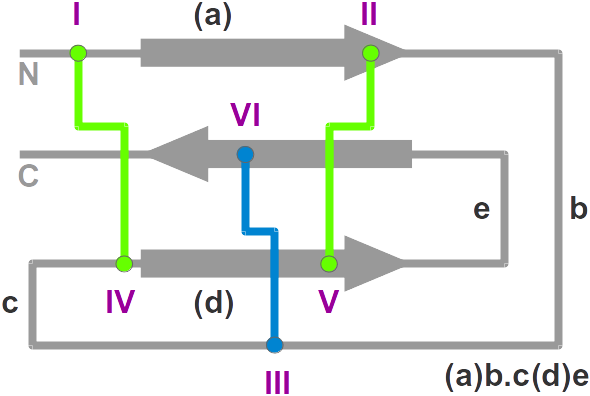
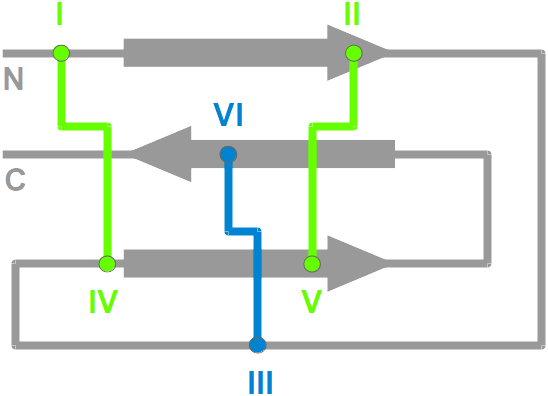
KNOTER3D determines if a three-dimensional structure is a knottin:
- New Protein Data Bank entries are filtered for
proteins with at least 3 disulfide bridges and a connectivity typical
of knottins (but not only), i.e. I-IV, II-V, III-VI..
- The selected protein structures are renumbered according to the
unique knottin numbering.
- The renumbered structures are
superimposed onto a reference knottin structure (the X-ray structure of
Cucurbita Pepo Trypsin Inhibitor II, CPTI-II, PDB ID 2btcI) for
residues 40, 60-61, 79-81 and 99-100. These residues approximately define
the Cystine-Stabilized
Beta-sheet (CSB) elementary motif in knottins that includes
the II-V and III-VI disulfide bridges.
Proteins that display both a CSB motif and a third I-IV disulfide bridge are indeed knottins.
- Root mean square deviations below 2.5 define knottins.
Ambiguous hits ( 2.5 < RMSD < 3.5 ) are manually checked.
If the RMSD is above 3.5, the structure is rejected.
KNOTER1D predicts if a protein sequence is a knottin
- The SwissProt/TrEMBL (UniProt) release is filtered for
cysteine-rich proteins. Each protein seq1 in the resulting subset is
compared to all known knottin sequences knot_seq2 using a similarity
score S based on:
- the BLAST P-value for the seq1/knot_seq2 comparison.
- the number of conserved cysteines of the knot when aligning seq1 onto
the multiple alignment of the family of knot_seq2.
- the compatibility of intercysteine loop lengths with known knottin
loop lengths.
- the taxonomic proximity between seq1 and knot_seq2.
- the similarity between the seq1 function and knottin functions
- the similarity between seq1 keywords and knottin keywords, and/or
dissimilarity between seq1 keywords and non-knottin keywords.
- New sequences are classified by decreasing S scores.
This list is then manually annotated for sequences to be included or
rejected.
General data for all selected knottin structures are
obtained from the Protein Data bank or MMDB and are stored in Table 1.
Local structural information is computed for each residue and stored in
Table 2.
Non-local information is computed for all interactions and stored in
Table 3.
General data for all selected knottin sequences are obtained from the
SwissProt/TrEMBL database and stored in Table 4.
|
| |
| Searching the database |
| |
| The button "Search
Database" points at a web form that allows to search either knottin
structures OR knottin sequences. |
| |
- Select the SEQUENCE or STRUCTURE database
- Make selections based on various criteria
- Family
Select one or all knottin protein families
- Keyword
Enter keyword (currently only one) to select entries that contain the
keyword in the Family, SwissProt ID, PDB ID, Descriptor, Source or
Function fields. The keyword is case insensitive.
exemples: 2eti, agatoxin, sodium, ITR2_momco, homo
- Method
allows selection of the method used to build the model (X-ray or NMR)
- Nomenclature
(i.e. loop lengths). Use Perl Regular expressions to select particular
loop lengths. For example, "^[2-4]$" will select loops with 2, 3, or 4
residues.
- Sort the results
- Family sort by family name
- SwissProt ID: sort by SwissProt ID
- PDB code: sort by PDB code
- Length: sort by sequence length
- RMS dev: sort by root mean square deviation from the reference structure 2btcI.
- Nomenclature: sort by the a-e loop lengths as used in the knottin
nomenclature
- Output tables
- The checkboxes allow selections for further sequence alignments or
structural superimpositions available via menus immediately above
and (possibly) below the output tables.
 The green buttons point at individual knottin cards
The green buttons point at individual knottin cards - The "Jmol" link in structure tables forwards you to 3D viewing of the structure with the Jmol Java applet.
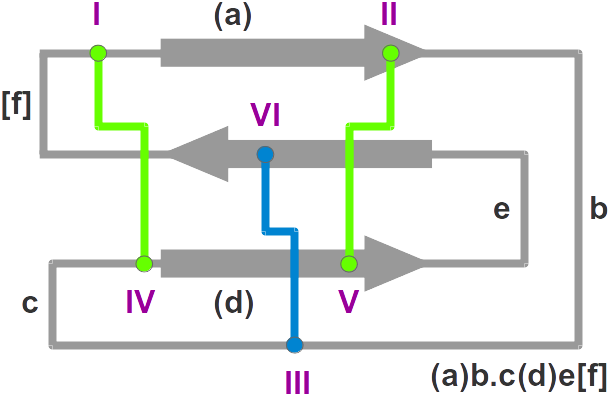
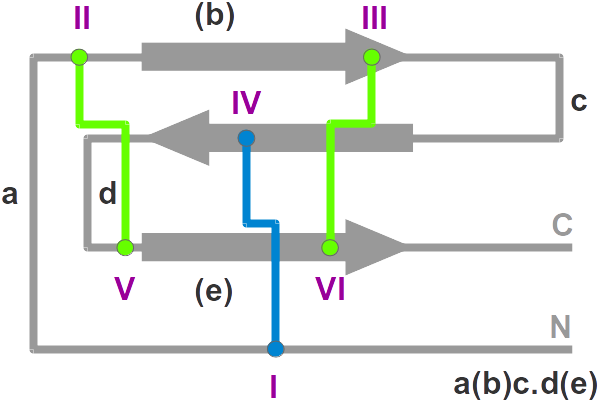
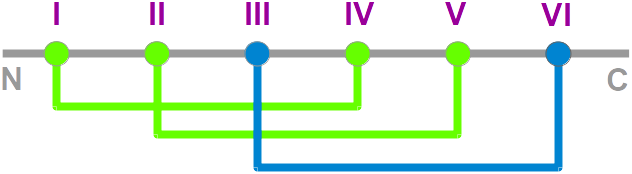
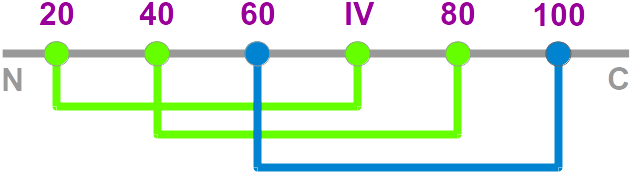
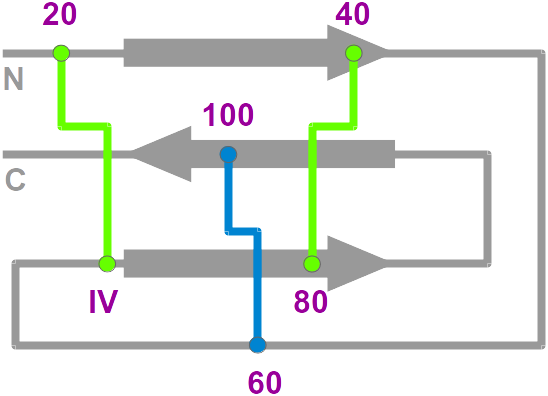
- Nomenclature: The knottin
nomenclature based on loop lengthes
- Family: The protein family
- SwissProt: The SwissProt ID ("-" means that no SwissProt ID is available yet)
- PDBsum: The PDB code points at the corresponding page in
PDBsum
- For 3D structures
R points at renumbered PDB files according to
the unique knottin numbering scheme
F
points at renumbered AND structurally fitted PDB files. The
renumbered structures are superimposed to a reference knottin structure
(the X-ray structure of Cucurbita Pepo Trypsin Inhibitor II, CPTI-II,
PDB ID 2btcI) for residues 40, 60-61, 79-81 and 99-100. These residues
define approximately the Cystine-Stabilized
Beta-sheet elementary motif in knottins that includes the
II-V and III-VI disulfide bridges
Chain:
The chain ID in the PDB file
Length:
The sequence length
RMS:
the RMS deviation from the reference structure for backbone atoms of
residues 40, 60-61, 79-81 and 99-100
Experimental:
The method for structure determination.
- Descriptor
- Source
- Tissue
- Function
- PubMed
- Individual knottin cards
General
data similar to those in the Main tabular output are available
Supplementary information essentially includes:
• A
"Collier de Perles"
two-dimensional representation. We gratefully thank Marie-Paule
Lefranc, for authorizing the use of the expression "Collier de Perles"
which originally refer to standardized 2D representations, in IMGT, the
international ImMunoGenetics information system® (http://imgt.cines.fr) [Lefranc,
M.-P.et al. Nucleic Acid. Res., 1999, 27, 209-212]
The original program for automatic drawing of Colliers de Perles was
rewritten and adapted to knottins by Quentin Kaas.
• For 3D
structures: a
sequence-structure table showing:
- Number:
The knottin unique numbering.
- Residue:
The amino acid at each position
- Secondary:
The secondary structure computed using the Stride
program.
- Phi:
The Phi angle computed using the Stride
program.
- Psi:
The Psi angle computed using the Stride
program.
- ASA:
The accessible surface area computed using the Stride
program
- PAC:
The percent of accessibility computed using the local PDBgeo program
- Yellow
columns indicate the
knotted disulfide bridges
Brown columns indicate
the additional disulfides
- Sequence alignments
- A "Get Alignment" button is available on top of the output tables.
- It is first necessary to select which knottins must be aligned using either
the "Select All" button or the checkboxes on the left side of each protein line.
- The renumbered sequences of the selected
proteins are passed to PAT
(Protein Analysis Toolkit written by Jerome Gracy jgracy@cbs.cnrs.fr)
that manages to produce and display an html color-coded
standardized alignment.
- Each alignment displayed on the KNOTTIN website is preceeded by a drop-down
menu that allows to:
- retrieve a text-formated version of the alignment in various formats for download
- create a sequence logo
- send the alignment to PAT
and switch the browser page to the PAT webserver for
more powerful analyses.
|
| BLASTing the database |
| |
| The button
"Blast Database" points at a web form that allows users to upload their
own sequence and BLAST (or search a HMM model database) this sequence against the KNOTTIN
database. |
| |
- Select search
method
BLAST: selecting
BLAST means that a blastp is performed against all sequences in the
database (including sequences of the PDB files)
HMM: selecting HMM means that a HMMsearch is performed against HMM models pre-computed for each family in
the database. The output diplays first the HHMsearch result, then the alignment of the user
sequence with the sequences used to build the matching model (HMMalign
is used for this).
- Paste sequence in FASTA format
The given sequence must conform to the
FASTA format
(see below) and must only contain permitted characters.
A sequence in FASTA format begins with a single-line
description, followed by lines of one-letter code sequence data.
- The description line starts with a greater than
symbol (">").
- The word following the greater than symbol
(">") immediately is the name of the sequence, the rest of the
line is the description.
- Name and description are optional.
- All lines of text should be shorter than 80
characters.
- The sequence ends if there is another greater
than symbol (">") at the beginning of a line.
Exemple:
>ITR2_ECBEL Trypsin Inhibitor II (EETI-II).
GCPRILMRCKQDSDCLAGCVCGPNGFCGSP
- Select MAX
number of hits
Restrict the number of hits shown in
the output
- Select MAX
p-value
Display only hits with below the
maximal selected p-value
- Submit
The user sequence is passed to PAT (Protein
Analysis Tools written by Jerome
Gracy) that manages to run BLASTA against the knottin
database.
The BLAST output is then HTML color-coded by PAT for display.
|
| CONFORMATION SEARCH |
| |
| The
button "CONFORMATION SEARCH" points at a web form that allows users to
search the KNOTTIN database for 20-residue long segments that satisfy
the selection explained below: |
| |
- Select display
Default display includes sequence and
position according to the unique
knottin numbering.
Users can choose to display the Phi and Psi angles, the secondary
structure and the percent of accessibility (PAC) by checking the
appropriate radio button.
All subsequent selections are optional,
although at least one selection must be performed. The maximum output
is currently limited to 1000 segments.
- Select protein
To select protein(s), type a list of
PDB IDs separated by blanks. (example "2btc 1ha9 1c6w").
The segment search will only be done on these proteins.
Note that if no other selection is performed, results will include all
sliding segments for each , i.e. as many segments as there is residues
in the protein. To avoid this, users can select a starting position for
the segment (see below)
- Starting position
To select only segments starting at a
given position, users can use the scrolling list. The position
correspond to the unique
knottin numbering. "*" means any position.
- Select sequence pattern
Users can select specific residue for
each position along the 20-residue segment using the scrolling lists.
"*" means any residue
- Select Ramachandran quadrant
Users can select the Ramachandran
quadrant for each residue along the 20-residue segment.
*: any quadrant
UL: Upper Left (Phi <0, Psi>0)
UR: Upper Right (Phi >0, Psi>0)
LL: Lower Left (Phi <0, Psi<0)
LR: Lower Right (Phi >0, Psi<0)
- Select secondary structure
Users can select the secondary
structure for each residue along the 20-residue segment. Secondary
structure is computed using the STRIDE program and uses the following
ciding:
*: any secondary structure
H: alpha-helix
G: 3-10 helix
I: PI-helix
E: Extended
B: Isolated bridge
T: Turn
C: Coil (none of the above)
- Output display
The
ouput consists of a succession of segments extracted from the KNOTTIN
database.
The first line indicates the protein (PDB ID, chain) and the sequence
of the segment.
The second line indicates the numbering of each residue according to
the knottin unique
numbering.
Subsequent lines indicates dihedral angles, percent of accessibility or
secondary structure, depending on the output request.
Output positions that match the requested selection are shown in red.
|
| TOOLS |
| |
The "Tools" menu points at a web form that allows users to submit a PDB file or a protein sequence.
KNOTER1D will determine if a 3D structure is a knottin
KNOTER3D will predict if a protein sequence is a knottin.
|
|
- Submit a protein structure or a protein sequence
The 3D structure must be in PDB format and the sequence in the FASTA format.
- Provide additional information if a sequence is submitted
KNOTER1D will search for homologs in the
SwissProt/TrEMBL database, and extract taxonomy, function or keywords
from the best homolog if any. The user has the possibility to
provide this information directly to KNOTER1D.
- Output
KNOTER3D or KNOTER1D will indicate if the protein is or is probably a knottin,
A detailed output of KNOTER1D is available via an html link.
If the protein is a knottin or putative knottin, standardized information is provided
• the original and standardized sequence numbers of the cysteines of the knot
• the knottin nomenclature
• a standardized Collier de Perles 2D representation
• a standardized renumbered alignment (knotted cysteines I, II, III, V and VI are renumbered 20, 40, 60, 80 and 100, respectively)
|
| Regular Expressions |
| |
Regular
expression can be used to select knottins in the KNOTTIN database
according to the knottin nomenclature,
i.e. according to loop lengths. To facilitate writing, separate
expressions are entered for each loop.
For example: ^1$ matches 1; 1 matches 1, 12, 31 etc; ^[2-4]$ matches 2,3 or 4; ^[1,4,7][0,1]*$ matches 1, 4, 7, 10, 40, 70, 11, 41, 71
Main metacharacters used in regular expressions are indicated below:
| character |
meaning |
example |
^ |
beginning of string |
^6 |
the string begins with "6" |
$ |
end of string |
6$ |
the string ends with "6" |
. |
any character (except newline) |
a.6 |
an "a" followed by any single character followed
by a "6" |
* |
match 0 or more times the preceding item |
c*3 |
any number of (or no) "c" followed by a "3" |
+ |
match 1 or more times the preceding item |
c+3 |
any number of "c" (at least one) followed by a
"3" |
? |
match 0 or 1 times the preceding item |
c?3 |
no or one "c" followed by a "3" |
[ ] |
set of characters |
[a,b,c]3 |
either "a" or "b" or "c" followed by a "3" |
| |
alternative |
^1|3$ |
either the string begins with a "1" or ends with
a "3" |
{ } |
repetition modifier |
c{2,5}3 |
two to five "c" followed by a "3" |
|